
Turning Back the Clock: A Retrospective Single-Blind Study on Brain Age Change in Response to Nutraceuticals Supplementation vs. Lifestyle Modifications

Abstract
:Death is […] not an absolute necessity essentially inherent in life itself.([1], p. 26)
1. Introduction
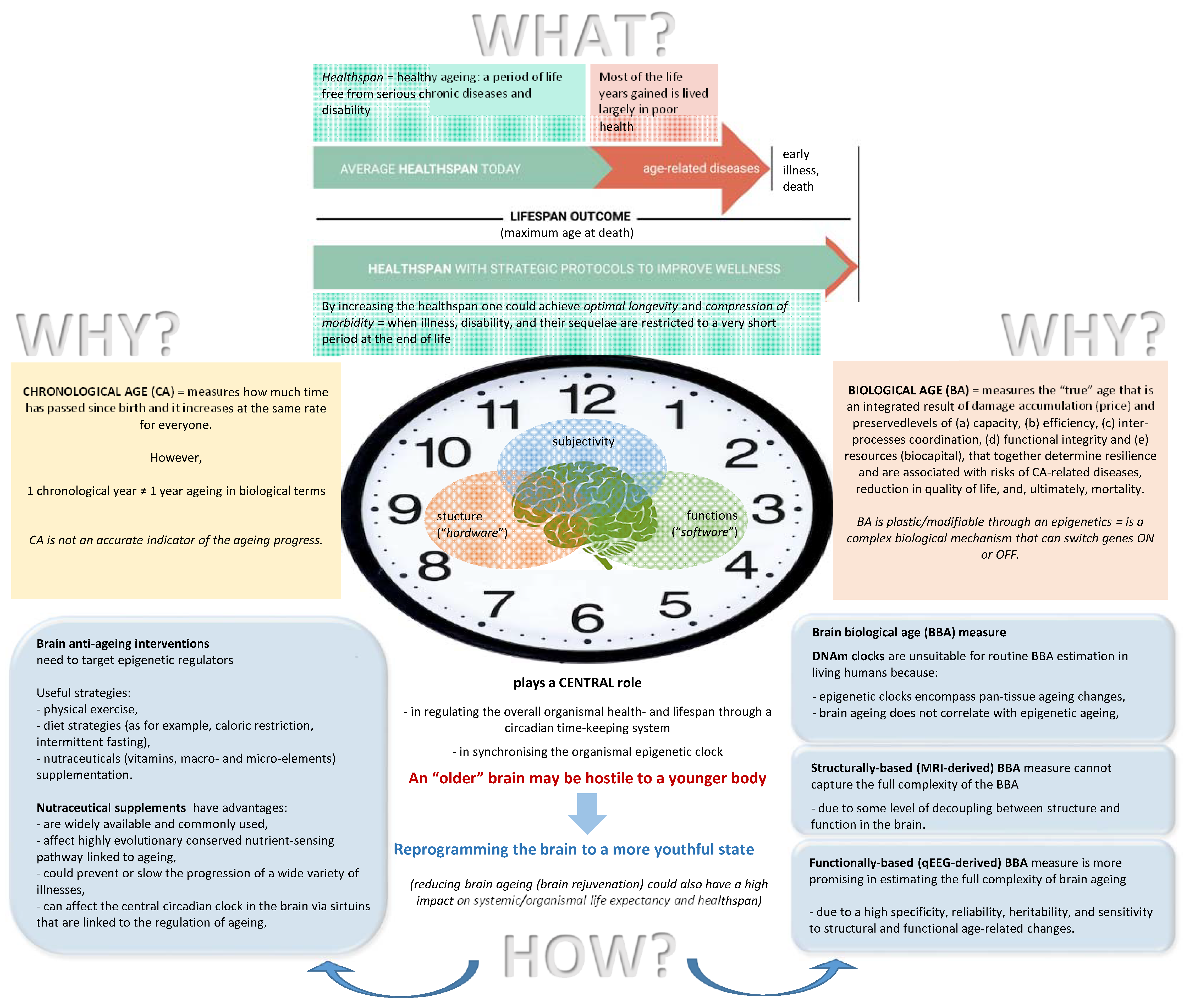
1.1. Brain Biological Age Estimation
- (a)
- (b)
- (c)
- It reflects both the brain’s structural characteristics (or “hardware”) such as the number of connections between neurons, fiber density, axonal diameter, degree of myelination and white matter integrity, as well as the integrity of the corticocortical and thalamocortical circuits, hippocampal volume (the hippocampus is a brain region central to both healthy memory function and also age-related memory decline [241]), number of active synapses in thalamic nuclei, brain hemodynamics and metabolism, and the number of potential neural pathways [231,242,243,244] and cognitive processes and functions (“neuropsychological competence” or “software”), such as memory performance, attention and processing speed, individual capacity for information processing (the capacity for storage, transfer, and retrieval of information) and cognitive preparedness (the brain’s capacity for higher-level cognitive functioning), network efficiency, and neural compensation at all ages, both in healthy individuals and in individuals with neurological conditions [245,246,247,248];
- (d)
- (e)
- (f)
1.2. Choosing a Brain Anti-Aging Intervention
1.3. Aim of the Study
2. Methods
2.1. Participants
2.2. EEG Recording and Acquisition
2.3. Estimation of Cerebral Physiological Age as a Proxy of the Brain’s BA—BBA
2.4. Interventions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Nutraceuticals | Lifestyle | p-Value | Test Type |
|---|---|---|---|---|
| Sample size (N) | 42 | 47 | Not applicable | Not applicable |
| Sex (% of females) | 73.8 | 53.2 | 0.00204 | Chi-square |
| Chronological age—CA (mean/st.d) | 54.1 (13) | 45.2 (7.3) | 0.00048 | Mann–Whitney U test |
| Brain biological age—BBA (mean/st.d) | 46.3 (11) | 37.7 (9.8) | 0.00042 | Mann–Whitney U test |
| Brain resources—BR (mean %/st.d) | 9.89 (20) | 8.99 (13) | Not significant | Mann–Whitney U test |
| Healthy lifestyle habits (% of those who have) | 16.7 | 12.8 | Not significant | Chi-square |
| Current health symptoms (% of those who have) | 33.3 | 40.2 | Not significant | Chi-square |
| Past health problems (% of those who had) | 64.3 | 57.4 | Not significant | Chi-square |
| Relatives with mind/brain disorders (% of those who have) | 16.7 | 23.4 | Not significant | Chi-square |
| Anxiety—Beck 1 (mean/st.d) | 8.2 (7.1) | 7.9 (6.5) | Not significant | Mann–Whitney U test |
| Anxiety—Ham 2 (mean/st.d) | 8.7 (6.6) | 8.6 (5.5) | Not significant | Mann–Whitney U test |
| Depression—Beck 3 (mean/st.d) | 6.2 (6.7) | 6.5 (4.8) | Not significant | Mann–Whitney U test |
| Big-5—neuroticism 4 (mean/st.d) | 2.8 (0.8) | 2.9 (0.7) | Not significant | Mann–Whitney U test |
| Handedness (% of right-handed) | 83.3 | 87.2 | Not significant | Chi-square |
| Marital status (% of married) | 73.8 | 83 | Not significant | Chi-square |
| Marital status (% of divorced) | 9.5 | 12.7 | Not significant | Chi-square |
| Marital status (% of single) | 16.7 | 4.3 | 0.002712 | Chi-square |
| Education (% of those who have a PhD) | 14.3 | 10.6 | Not significant | Chi-square |
| Education (% of those who graduated from university or institute) | 69 | 74.4 | Not significant | Chi-square |
| Education (% of those who completed high school (≥11–12 years)) | 16.7 | 15 | Not significant | Chi-square |
| Job (% of directors or CEOs) | 21.4 | 17 | Not significant | Chi-square |
| Job (% of senior managers) | 38.1 | 38.3 | Not significant | Chi-square |
| Job (% of junior managers) | 35.7 | 38.3 | Not significant | Chi-square |
| Job (% of students or trainees) | 4.8 | 6.4 | Not significant | Chi-square |
| Number of interests or hobbies (mean/st.d) | 4.3 (1.8) | 3.6 (1.6) | 0.0394 | Mann–Whitney U test |
| Smoking (% of those who smoke) | 7.1 | 2.1 | Not significant | Chi-square |
| Alcohol consumption (1–2 drinks * per week; %) | 40.5 | 40.4 | Not significant | Chi-square |
| Alcohol consumption (3–4 drinks per week; %) | 47.6 | 38.3 | Not significant | Chi-square |
| Alcohol consumption (5–7 drinks per week; %) | 7.1 | 8.5 | Not significant | Chi-square |
| Alcohol consumption (8–10 drinks per week; %) | 4.8 | 12.8 | 0.04808 | Chi-square |
2.5. Statistical Analyses
3. Results
3.1. Demographic Characteristics
3.2. Neurophysiological Findings: BBA and BR
3.3. Psychometrics and Health Symptoms
4. Discussion
5. Conclusions, Significance, Limitations, and Future Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weissman, A. Essays Upon Heredity and Kindred Biological Problems, 2nd ed.; Clarendon Press: Oxford, UK, 1891. [Google Scholar]
- Cave, S. Immortality: The Quest to Live Forever and How It Drives Civilization; Crown Publishers: New York, NY, USA, 2012. [Google Scholar]
- Cholbi, M.J. The Science of Immortality; John Templeton Foundation: West Conshohocken, PA, USA, 2018. [Google Scholar]
- Bostrom, N. Are you living in a computer simulation? Philos. Q. 2003, 53, 243–255. [Google Scholar] [CrossRef]
- Bostrom, N. The Future of Humanity. In New Waves in Philosophy of Technology; Berg Olsen, J.-K., Selinger, E., Aldershot, S.R., Eds.; Palgrave McMillan: New York, NY, USA, 2009; pp. 186–216. [Google Scholar]
- Hughes, J.J. The compatibility of religious and transhumanist views of metaphysics, suffering, virtue and transcendence in an enhanced future. Glob. Spiral 2007, 8, 1–40. Available online: https://ieet.org/archive/20070326-Hughes-ASU-H+Religion.pdf (accessed on 14 February 2022).
- Fingelkurts, A.A.; Fingelkurts, A.A. After human. Futura 2018, 4, 60–74. [Google Scholar]
- Zhavoronkov, A. The Ageless Generation: How Advances in Biomedicine will Transform the Global Economy; St. Martin’s Press: New York, NY, USA, 2013. [Google Scholar]
- Sinclair, D.A. Lifespan: Why We Age—And Why We Don’t Have To; Thorsons: London, UK, 2019. [Google Scholar]
- West, M.D.; Sternberg, H.; Labat, I.; Janus, J.; Chapman, K.B.; Malik, N.N.; De Grey, A.D.; Larocca, D. Toward a unified theory of aging and regeneration. Regen. Med. 2019, 14, 867–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barzilai, N. Age Later: Health Span, Life Span, and the New Science of Longevity; St Martin’s Press: London, UK, 2020. [Google Scholar]
- Scott, A.J.; Ellison, M.; Sinclair, D.A. The economic value of targeting aging. Nat. Aging 2021, 1, 616–623. [Google Scholar] [CrossRef]
- de Grey, A.D.N.J. A thousand years young. Futurist 2012, 46, 18–23. [Google Scholar]
- de Grey, A.D.N.J. Escape Velocity: Why the Prospect of Extreme Human Life Extension Matters Now. PLoS Biol. 2004, 2, e187. [Google Scholar] [CrossRef] [Green Version]
- Oeppen, J.; Vaupel, J.W. Broken limits to life expectancy. Science 2002, 296, 1029–1031. [Google Scholar] [CrossRef] [Green Version]
- Zhavoronkov, A.; Bischof, E.; Lee, K.-F. Artificial intelligence in longevity medicine. Nat. Aging 2021, 1, 5–7. [Google Scholar] [CrossRef]
- GBD; Demographics Collaborators. Global age-sex-specific fertility, mortality, healthy life expectancy (HALE), and population estimates in 204 countries and territories, 1950–2019: A comprehensive demographic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1160–1203. [Google Scholar] [CrossRef]
- Olshansky, S.J. From Lifespan to Healthspan. JAMA 2018, 320, 1323–1324. [Google Scholar] [CrossRef]
- Cutler, R.G.; Mattson, M.P. The adversities of ageing. Ageing Res. Rev. 2006, 5, 221–238. [Google Scholar] [CrossRef] [PubMed]
- de Grey, A.D. Life Span Extension Research and Public Debate: Societal Considerations. Stud. Ethic Law, Technol. 2007, 1. [Google Scholar] [CrossRef]
- Seals, D.R.; Melov, S. Translational Geroscience: Emphasizing function to achieve optimal longevity. Aging 2014, 6, 718–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fries, J.F. Aging, natural death, and the compression of morbidity. N. Engl. J. Med. 1980, 303, 130–135. [Google Scholar] [CrossRef] [Green Version]
- Omran, A.R. The Epidemiologic Transition: A Theory of the Epidemiology of Population Change. Milbank Mem. Fund Q. 1971, 49, 509. [Google Scholar] [CrossRef]
- Olshansky, S.J.; Ault, A.B. The Fourth Stage of the Epidemiologic Transition: The Age of Delayed Degenerative Diseases. Milbank Q. 1986, 64, 355. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J.; Kapahi, P.; Lithgow, G.J.; Melov, S.; Newman, J.C.; Verdin, E. From discoveries in ageing research to therapeutics for healthy ageing. Nature 2019, 571, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P. What is “phenoptosis” and how to fight it? Biochemistry 2012, 77, 689–706. [Google Scholar] [CrossRef]
- Moskalev, A.A.; Shaposhnikov, M.V.; Plyusnina, E.N.; Zhavoronkov, A.; Budovsky, A.; Yanai, H.; Fraifeld, V.E. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res. Rev. 2013, 12, 661–684. [Google Scholar] [CrossRef]
- Sierra, F. Geroscience and the challenges of aging societies. Aging Med. 2019, 2, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Galkin, F.; Mamoshina, P.; Aliper, A.; de Magalhães, J.P.; Gladyshev, V.N.; Zhavoronkov, A. Biohorology and biomarkers of aging: Current state-of-the-art, challenges and opportunities. Ageing Res. Rev. 2020, 60, 101050. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Aging is a specific biological function rather than the result of a disorder in complex living systems: Biochemical evidence in support of Weismann’s hypothesis. Biochemistry 1997, 62, 1191–1195. [Google Scholar]
- de Grey, A.D.N.J. Challenging but essential targets for genuine antiageing drugs. Expert Opin. Ther. Targets 2003, 7, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Butler, R.N.; Sprott, R.; Warner, H.; Bland, J.; Feuers, R.; Forster, M.; Fillit, H.; Harman, S.M.; Hewitt, M.; Hyman, M.; et al. Aging: The Reality: Biomarkers of Aging: From Primitive Organisms to Humans. J. Gerontol. Ser. A 2004, 59, B560–B567. [Google Scholar] [CrossRef] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking Aging to Chronic Disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef] [Green Version]
- Jylhävä, J.; Pedersen, N.L.; Hägg, S. Biological Age Predictors. Ebiomedicine 2017, 21, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Gems, D.; de Magalhães, J.P. The hoverfly and the wasp: A critique of the hallmarks of aging as a paradigm. Ageing Res. Rev. 2021, 70, 101407. [Google Scholar] [CrossRef]
- Jia, L.; Zhang, W.; Chen, X. Common methods of biological age estimation. Clin. Interv. Aging 2017, 12, 759–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, D.; Sternäng, O.; Wahlin, Å. Genetic and Environmental Influences on Longitudinal Trajectories of Functional Biological Age: Comparisons Across Gender. Behav. Genet. 2017, 47, 375–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gott, A.; Andrews, C.; Larriva-Hormigos, M.; Spencer, K.; Bateson, M.; Nettle, D. Chronological age, biological age, and individual variation in the stress response in the European starling: A follow-up study. PeerJ 2018, 6, e5842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunn, D.A.; Rexbye, H.; Griffiths, C.; Murray, P.G.; Fereday, A.; Catt, S.D.; Tomlin, C.C.; Strongitharm, B.H.; Perrett, D.; Catt, M.; et al. Why Some Women Look Young for Their Age. PLoS ONE 2009, 4, e8021. [Google Scholar] [CrossRef] [Green Version]
- Ahadi, S.; Zhou, W.; Rose, S.M.S.-F.; Sailani, M.R.; Contrepois, K.; Avina, M.; Ashland, M.; Brunet, A.; Snyder, M. Personal aging markers and ageotypes revealed by deep longitudinal profiling. Nat. Med. 2020, 26, 83–90. [Google Scholar] [CrossRef]
- Lehallier, B.; Gate, D.; Schaum, N.; Nanasi, T.; Lee, S.E.; Yousef, H.; Losada, P.M.; Berdnik, D.; Keller, A.; Verghese, J.; et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat. Med. 2019, 25, 1843–1850. [Google Scholar] [CrossRef]
- Mamoshina, P.; Kochetov, K.; Cortese, F.; Kovalchuk, A.; Aliper, A.; Putin, E.; Scheibye-Knudsen, M.; Cantor, C.R.; Skjodt, N.M.; Kovalchuk, O.; et al. Blood Biochemistry Analysis to Detect Smoking Status and Quantify Accelerated Aging in Smokers. Sci. Rep. 2019, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- Jagust, W.J. Youthfulness begins in youth. Nat. Aging 2021, 1, 239–240. [Google Scholar] [CrossRef]
- Levine, M.E. Modeling the rate of senescence: Can estimated biological age predict mortality more accurately than chronological age? J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.G.; Suh, E.; Lee, J.-W.; Kim, D.W.; Cho, K.H.; Bae, C.-Y. Biological age as a health index for mortality and major age-related disease incidence in Koreans: National Health Insurance Service—Health screening 11-year follow-up study. Clin. Interv. Aging 2018, 13, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Zhavoronkov, A.; Mamoshina, P. Deep Aging Clocks: The Emergence of AI-Based Biomarkers of Aging and Longevity. Trends Pharmacol. Sci. 2019, 40, 546–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhavoronkov, A.; Li, R.; Ma, C.; Mamoshina, P. Deep biomarkers of aging and longevity: From research to applications. Aging 2019, 11, 10771–10780. [Google Scholar] [CrossRef] [PubMed]
- Borkan, G.A.; Norris, A.H. Assessment of Biological Age Using a Profile of Physical Parameters. J. Gerontol. 1980, 35, 177–184. [Google Scholar] [CrossRef]
- Jackson, S.H.; Weale, M.R.; Weale, R.A. Biological age-what is it and can it be measured? Arch. Gerontol. Geriatr. 2003, 36, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Fried, L.P.; Xue, Q.-L.; Cappola, A.R.; Ferrucci, L.; Chaves, P.; Varadhan, R.; Guralnik, J.M.; Leng, S.X.; Semba, R.D.; Walston, J.D.; et al. Nonlinear Multisystem Physiological Dysregulation Associated With Frailty in Older Women: Implications for Etiology and Treatment. J. Gerontol. Ser. A 2009, 64A, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, K.; Gaser, C. Longitudinal changes in individual BrainAGE in healthy ageing, mild cognitive impairment, and Alzheimer’s Disease. GeroPsych 2012, 25, 235–245. [Google Scholar] [CrossRef]
- Cole, J.H.; Annus, T.; Wilson, L.R.; Remtulla, R.; Hong, Y.T.; Fryer, T.D.; Acosta-Cabronero, J.; Cardenas-Blanco, A.; Smith, R.; Menon, D.K. Brain predicted age in Down syndrome is associated with beta amyloid deposition and cognitive decline. Neurobiol. Ageing 2017, 56, 41–49. [Google Scholar] [CrossRef]
- Cole, J.H.; Ritchie, S.J.; Bastin, M.E.; Hernández, M.C.V.; Maniega, S.M.; Royle, N.; Corley, J.; Pattie, A.; Harris, S.E.; Zhang, Q.; et al. Brain age predicts mortality. Mol. Psychiatry 2018, 23, 1385–1392. [Google Scholar] [CrossRef] [Green Version]
- Christensen, K.; Wojczynski, M.K.; Pedersen, J.K.; Larsen, L.A.; Kløjgaard, S.; Skytthe, A.; McGue, M.; Vaupel, J.W.; Province, M.A. Mechanisms underlying familial aggregation of exceptional health and survival: A three-generation cohort study. Aging Cell 2020, 19, 13228. [Google Scholar] [CrossRef]
- Stern, Y.; Gurland, B.; Tatemichi, T.K.; Tang, M.X.; Wilder, D.; Mayeux, R. Influence of education and occupation on the incidence of Alzheimer’s disease. JAMA 1994, 271, 1004–1010. [Google Scholar] [CrossRef]
- Sayer, A.A.; Cooper, C.; Evans, J.R.; Rauf, A.; Wormald, R.P.; Osmond, C.; Barker, D.J. Are rates of ageing determined in utero? Age Ageing 1998, 27, 579–583. [Google Scholar] [CrossRef] [Green Version]
- Syddall, H.E.; Sayer, A.A.; Simmonds, S.J.; Osmond, C.; Cox, V.; Dennison, E.M.; Barker, D.J.; Cooper, C. Birth weight, infant weight gain, and cause-specific mortality: The Hertfordshire Cohort Study. Am. J. Epidemiol. 2005, 161, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Glatt, S.J.; Chayavichitsilp, P.; Depp, C.; Schork, N.J.; Jeste, D.V. Successful Aging: From Phenotype to Genotype. Biol. Psychiatry 2007, 62, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Pruchno, R.A.; Wilson-Genderson, M.; Rose, M.; Cartwright, F. Successful Aging: Early Influences and Contemporary Characteristics. Gerontologist 2010, 50, 821–833. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.H.; Ferraro, K.F. Childhood Misfortune as a Threat to Successful Aging: Avoiding Disease. Gerontologist 2012, 52, 111–120. [Google Scholar] [CrossRef]
- Wang, H.-X.; Gustafson, D.R.; Kivipelto, M.; Pedersen, N.L.; Skoog, I.; Windblad, B.; Fratiglioni, L. Education halves the risk of dementia due to apolipoprotein ε4 allele: A collaborative study from the Swedish Brain Power initiative. Neurobiol. Aging 2012, 33, 1007.e1–1007.e7. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.M.; Peiffer, J.J.; Taddei, K.; Lui, J.K.; Laws, S.M.; Gupta, V.B.; Taddei, T.; Ward, V.K.; Rodrigues, M.A.; Burnham, S.; et al. Physical activity and amyloid-beta plasma and brain levels: Results from the Australian imageing, biomarkers and lifestyle study of ageing. Mol. Psychiatry 2013, 18, 875–881. [Google Scholar] [CrossRef] [Green Version]
- Sexton, C.E.; Storsve, A.B.; Walhovd, K.B.; Johansen-Berg, H.; Fjell, A.M. Poor sleep quality is associated with increased cortical atrophy in community-dwelling adults. Neurology 2014, 83, 967–973. [Google Scholar] [CrossRef] [Green Version]
- Arenaza-Urquijo, E.M.; Gonneaud, J.; Fouquet, M.; Perrotin, A.; Mezenge, F.; Landeau, B.; Egret, S.; De la Sayette, V.; Desgranges, B.; Chételat, G. Interaction between years of education and APOE epsilon4 status on frontal and temporal metabolism. Neurology 2015, 85, 1392–1399. [Google Scholar] [CrossRef] [Green Version]
- Gardener, S.L.; Rainey-Smith, S.R.; Barnes, M.B.; Sohrabi, H.R.; Weinborn, M.; Lim, Y.Y.; Harrington, K.; Taddei, K.; Gu, Y.; Rembach, A.; et al. Dietary patterns and cognitive decline in an Australian study of ageing. Mol. Psychiatry 2015, 20, 860–866. [Google Scholar] [CrossRef]
- Branger, P.; Arenaza-Urquijo, E.M.; Tomadesso, C.; Mézenge, F.; André, C.; de Flores, R.; Mutlu, J.; de La Sayette, V.; Eustache, F.; Chételat, G.; et al. Relationships between sleep quality and brain volume, metabolism, and amyloid deposition in late adulthood. Neurobiol. Aging 2016, 41, 107–114. [Google Scholar] [CrossRef]
- Löwe, L.C.; Gaser, C.; Franke, K.; Alzheimer’s Disease Neuroimaging Initiative. The Effect of the APOE Genotype on Individual BrainAGE in Normal Aging, Mild Cognitive Impairment, and Alzheimer’s Disease. PLoS ONE 2016, 11, e0157514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffener, J.; Habeck, C.; O’Shea, D.; Razlighi, Q.; Bherer, L.; Stern, Y. Differences between chronological and brain age are related to education and self-reported physical activity. Neurobiol. Aging 2016, 40, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vemuri, P.; Lesnick, T.G.; Przybelski, S.A.; Knopman, D.S.; Machulda, M.; Lowe, V.J.; Mielke, M.M.; Roberts, R.O.; Gunter, J.L.; Senjem, M.L.; et al. Effect of intellectual enrichment on AD biomarker trajectories: Longitudinal imageing study. Neurology 2016, 86, 1128–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reutrakul, S.; Van Cauter, E. Sleep influences on obesity, insulin resistance, and risk of type 2 diabetes. Metab. Clin. Exp. 2018, 84, 56–66. [Google Scholar] [CrossRef]
- Hayflick, L. Biological Aging Is No Longer an Unsolved Problem. Ann. N. Y. Acad. Sci. 2007, 1100, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Larocca, D.; Lee, J.; West, M.; Labat, I.; Sternberg, H. No Time to Age: Uncoupling Aging from Chronological Time. Genes 2021, 12, 611. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The Mammalian Epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar] [CrossRef]
- Griñán-Ferré, C.; Bellver-Sanchis, A.; Izquierdo, V.; Corpas, R.; Roig-Soriano, J.; Chillón, M.; Andres-Lacueva, C.; Somogyvári, M.; Sőti, C.; Sanfeliu, C.; et al. The pleiotropic neuroprotective effects of resveratrol in cognitive decline and Alzheimer’s disease pathology: From antioxidant to epigenetic therapy. Ageing Res. Rev. 2021, 67, 101271. [Google Scholar] [CrossRef]
- van Otterdijk, S.D.; Michels, K.B. Transgenerational epigenetic inheritance in mammals: How good is the evidence? FASEB J. 2016, 30, 2457–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trerotola, M.; Relli, V.; Simeone, P.; Alberti, S. Epigenetic inheritance and the missing heritability. Hum. Genom. 2015, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, S.; Lin, X.-H.; Xiong, Y.-M.; Liu, M.-E.; Yu, T.-T.; Lv, M.; Zhao, W.; Xu, G.-F.; Ding, G.-L.; Xu, C.-M.; et al. Prevalence of Prediabetes Risk in Offspring Born to Mothers with Hyperandrogenism. Ebiomedicine 2017, 16, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo, V.; Palomera-Ávalos, V.; López-Ruiz, S.; Canudas, A.-M.; Pallàs, M.; Griñán-Ferré, C. Maternal Resveratrol Supplementation Prevents Cognitive Decline in Senescent Mice Offspring. Int. J. Mol. Sci. 2019, 20, 1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumey, L. Decreased birthweights in infants after maternal in utero exposure to the Dutch famine of 1944–1945. Paediatr. Périnat. Epidemiology 1992, 6, 240–253. [Google Scholar] [CrossRef]
- Lumey, L.H.; Stein, A.; Kahn, H.; Bruin, K.M.V.D.P.-D.; Blauw, G.J.; Zybert, P.A.; Susser, E.S. Cohort Profile: The Dutch Hunger Winter Families Study. Leuk. Res. 2007, 36, 1196–1204. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, J.; Sandboge, S.; Salonen, M.K.; Kajantie, E.; Osmond, C. Long-term consequences of maternal overweight in pregnancy on offspring later health: Findings from the Helsinki Birth Cohort Study. Ann. Med. 2014, 46, 434–438. [Google Scholar] [CrossRef]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef] [Green Version]
- Guillaumet-Adkins, A.; Yañez, Y.; Peris-Diaz, M.D.; Calabria, I.; Palanca-Ballester, C.; Sandoval, J. Epigenetics and Oxidative Stress in Aging. Oxidative Med. Cell. Longev. 2017, 2017, 9175806. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.; Simpkins, J.W.; Ji, X.; Leis, M.; Stambler, I. The Critical Need to Promote Research of Aging and Aging-related Diseases to Improve Health and Longevity of the Elderly Population. Aging Dis. 2014, 6, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, A.E.; Sinclair, D.A. Epigenetic changes during aging and their reprogramming potential. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 61–83. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.N.; Hodges, R.; Hanes, D.; Stack, E.; Cheishvili, D.; Szyf, M.; Henkel, J.; Twedt, M.W.; Giannopoulou, D.; Herdell, J.; et al. Potential reversal of epigenetic age using a diet and lifestyle intervention: A pilot randomized clinical trial. Aging 2021, 13, 9419–9432. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, H.; Chapman, K.; Guigova, A.; Teichroeb, J.; Lacher, M.D.; Sternberg, H.; Singec, I.; Briggs, L.; Wheeler, J.; Sampathkumar, J.; et al. Spontaneous reversal of the developmental aging of normal human cells following transcriptional reprogramming. Regen. Med. 2010, 5, 345–363. [Google Scholar] [CrossRef]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Aït-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.-M.; De Vos, J.; et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef] [Green Version]
- Wahlestedt, M.; Erlandsson, E.; Kristiansen, T.; Lu, R.; Brakebusch, C.; Weissman, I.L.; Yuan, J.; Martin-Gonzalez, J.; Bryder, D. Clonal reversal of ageing-associated stem cell lineage bias via a pluripotent intermediate. Nat. Commun. 2017, 8, 14533. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Bignone, P.A.; Coles, L.S.; Liu, Y.; Snyder, E.; Larocca, D. Induced pluripotency and spontaneous reversal of cellular aging in supercentenarian donor cells. Biochem. Biophys. Res. Commun. 2020, 525, 563–569. [Google Scholar] [CrossRef]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S.; et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 2020, 588, 124–129. [Google Scholar] [CrossRef]
- Johnson, A.A.; Akman, K.; Calimport, S.R.G.; Wuttke, D.; Stolzing, A.; de Magalhães, J.P. The Role of DNA Methylation in Aging, Rejuvenation, and Age-Related Disease. Rejuv. Res. 2012, 15, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Mitteldorf, J.J. How does the body know how old it is? Introducing the epigenetic clock hypothesis. Biochemistry 2013, 78, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Field, A.E.; Robertson, N.A.; Wang, T.; Havas, A.; Ideker, T.; Adams, P.D. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol. Cell 2018, 71, 882–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wei, W.; Zhao, Q.-Y.; Widagdo, J.; Baker-Andresen, D.; Flavell, C.R.; D’Alessio, A.; Zhang, Y.; Bredy, T.W. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc. Natl. Acad. Sci. USA 2014, 111, 7120–7125. [Google Scholar] [CrossRef] [Green Version]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Ritchie, S.J.; Muniz-Terrera, G.; Harris, S.E.; Gibson, J.; Redmond, P.; Cox, S.R.; Pattie, A.; et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int. J. Epidemiol. 2015, 44, 1388–1396. [Google Scholar] [CrossRef] [Green Version]
- Maier, H.; Gampe, J.; Jeune, B.; Robine, J.-M.; Vaupel, J.W. (Eds.) Supercentenarians; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Guevara, E.E.; Lawler, R.R.; Staes, N.; White, C.M.; Sherwood, C.C.; Ely, J.J.; Hopkins, W.D.; Bradley, B.J. Age-associated epigenetic change in chimpanzees and humans. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190616. [Google Scholar] [CrossRef]
- Skulachev, V.P.; Holtze, S.; Vyssokikh, M.Y.; Bakeeva, L.E.; Skulachev, M.V.; Markov, A.V.; Hildebrandt, T.B.; Sadovnichii, V.A. Neoteny, prolongation of youth: From naked mole rats to “naked apes” (Humans). Physiol. Rev. 2017, 97, 699–720. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Pirazzini, C.; Bacalini, M.G.; Gentilini, D.; Di Blasio, A.M.; Delledonne, M.; Mari, D.; Arosio, B.; Monti, D.; Passarino, G.; et al. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging 2015, 7, 1159–1170. [Google Scholar] [CrossRef] [Green Version]
- Gutman, D.; Rivkin, E.; Fadida, A.; Sharvit, L.; Hermush, V.; Rubin, E.; Kirshner, D.; Sabin, I.; Dwolatzky, T.; Atzmon, G. Exceptionally Long-Lived Individuals (ELLI) Demonstrate Slower Aging Rate Calculated by DNA Methylation Clocks as Possible Modulators for Healthy Longevity. Int. J. Mol. Sci. 2020, 21, 615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fransquet, P.D.; Wrigglesworth, J.; Woods, R.L.; Ernst, M.E.; Ryan, J. The epigenetic clock as a predictor of disease and mortality risk: A systematic review and meta-analysis. Clin. Epigenetics 2019, 11, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grodstein, F.; Lemos, B.; Yu, L.; Iatrou, A.; De Jager, P.L.; Bennett, D.A. Characteristics of Epigenetic Clocks Across Blood and Brain Tissue in Older Women and Men. Front. Neurosci. 2021, 14, 555307. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.S.; Lloyd-Jones, D.M.; O’Flaherty, M.; Capewell, S.; Kershaw, K.N.; Carnethon, M.; Khan, S. Trends in Cardiometabolic Mortality in the United States, 1999-2017. JAMA 2019, 322, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Beck, J.C.; Bower, J.H.; Roberts, E.; Ritz, B.; Ross, G.W.; Abbott, R.D.; Savica, R.; Van Den Eeden, S.K.; Willis, A.W.; et al. Prevalence of Parkinson’s disease across North America. NPJ Parkinson’s Dis. 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Moya-Alvarado, G.; Gershoni-Emek, N.; Perlson, E.; Bronfman, F.C. Neurodegeneration and alzheimer’s disease (AD). What can proteomics tell us about the alzheimer’s brain? Mol. Cell Proteom. 2016, 15, 409–425. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Wang, Q.; Chung, S.K.; Shen, J. Crosstalk of metabolic factors and neurogenic signaling in adult neurogenesis: Implication of metabolic regulation for mental and neurological diseases. Neurochem. Int. 2017, 106, 24–36. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, B.J.; Kaeberlein, M. An energetics perspective on geroscience: Mitochondrial protonmotive force and aging. Geroscience 2021, 43, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Harper, S. Economic and social implications of aging societies. Science 2014, 346, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Patton, A.P.; Hastings, M.H. The suprachiasmatic nucleus. Curr. Biol. 2018, 28, R816–R822. [Google Scholar] [CrossRef] [Green Version]
- Hastings, M.; Maywood, E.; Brancaccio, M. The Mammalian Circadian Timing System and the Suprachiasmatic Nucleus as Its Pacemaker. Biology 2019, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Sinturel, F.; Gos, P.; Petrenko, V.; Hagedorn, C.; Kreppel, F.; Storch, K.-F.; Knutti, D.; Liani, A.; Weitz, C.; Emmenegger, Y.; et al. Circadian hepatocyte clocks keep synchrony in the absence of a master pacemaker in the suprachiasmatic nucleus or other extrahepatic clocks. Genes Dev. 2021, 35, 329–334. [Google Scholar] [CrossRef]
- Berger, J. Regulation of circadian rhythms. J. Appl. Biomed. 2004, 2, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Mohawk, J.A.; Green, C.B.; Takahashi, J.S. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 2012, 35, 445–462. [Google Scholar] [CrossRef] [Green Version]
- Astiz, M.; Heyde, I.; Oster, H. Mechanisms of Communication in the Mammalian Circadian Timing System. Int. J. Mol. Sci. 2019, 20, 343. [Google Scholar] [CrossRef] [Green Version]
- Bass, J. Circadian topology of metabolism. Nature 2012, 491, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Lee, C.C. The circadian clock: Pacemaker and tumour suppressor. Nat. Rev. Cancer 2003, 3, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Sahar, S.; Sassone-Corsi, P. Metabolism and cancer: The circadian clock connection. Nat. Rev. Cancer 2009, 9, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Froy, O. Circadian Rhythms, Aging, and Life Span in Mammals. Physiology 2011, 26, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Kondratova, A.A.; Kondratov, R.V. The circadian clock and pathology of the ageing brain. Nat. Rev. Neurosci. 2012, 13, 325–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masri, S. Sirtuin-dependent clock control: New advances in metabolism, aging and cancer. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samoilova, E.M.; Belopasov, V.V.; Ekusheva, E.V.; Zhang, C.; Troitskiy, A.V.; Baklaushev, V.P. Epigenetic Clock and Circadian Rhythms in Stem Cell Aging and Rejuvenation. J. Pers. Med. 2021, 11, 1050. [Google Scholar] [CrossRef]
- Lehmann, M.; Canatelli-Mallat, M.; Chiavellini, P.; Goya, R.G. A hierarchical model for the control of epigenetic aging in mammals. Ageing Res. Rev. 2020, 62, 101134. [Google Scholar] [CrossRef]
- Paixao, L.; Sikka, P.; Sun, H.; Jain, A.; Hogan, J.; Thomas, R.; Westover, M.B. Excess brain age in the sleep electroencephalogram predicts reduced life expectancy. Neurobiol. Aging 2020, 88, 150–155. [Google Scholar] [CrossRef]
- Elliott, M.L.; Belsky, D.W.; Knodt, A.R.; Ireland, D.; Melzer, T.R.; Poulton, R.; Ramrakha, S.; Caspi, A.; Moffitt, T.E.; Hariri, A.R. Brain-age in midlife is associated with accelerated biological aging and cognitive decline in a longitudinal birth cohort. Mol. Psychiatry 2021, 26, 3829–3838. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.H.; Marioni, R.E.; Harris, S.E.; Deary, I.J. Brain age and other bodily ‘ages’: Implications for neuropsychiatry. Mol. Psychiatry 2019, 24, 266–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nenadić, I.; Dietzek, M.; Langbein, K.; Sauer, H.; Gaser, C. BrainAGE score indicates accelerated brain aging in schizophrenia, but not bipolar disorder. Psychiatry Res. Neuroimaging 2017, 266, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Pardoe, H.R.; Cole, J.H.; Blackmon, K.; Thesen, T.; Kuzniecky, R. Structural brain changes in medically refractory focal epilepsy resemble premature brain aging. Epilepsy Res. 2017, 133, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Higgins-Chen, A.T.; Boks, M.P.; Vinkers, C.H.; Kahn, R.S.; Levine, M.E. Schizophrenia and Epigenetic Aging Biomarkers: Increased Mortality, Reduced Cancer Risk, and Unique Clozapine Effects. Biol. Psychiatry 2020, 88, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Ori, A.P.S.; Olde Loohuis, L.M.; Guintivano, J.; Hannon, E.; Dempster, E.; St. Clair, D.; Bass, N.J.; McQuillin, A.; Mill, J.; Sullivan, P.F.; et al. Epigenetic age is accelerated in schizophrenia with age- and sex-specific effects and associated with polygenic disease risk. bioRxiv 2021, 727859. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Aghajani, M.; Clark, S.L.; Chan, R.; Hattab, M.W.; Shabalin, A.A.; Zhao, M.; Kumar, G.; Xie, L.Y.; Jansen, R.; et al. Epigenetic Aging in Major Depressive Disorder. Am. J. Psychiatry 2018, 175, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Protsenko, E.; Yang, R.; Nier, B.; Reus, V.; Hammamieh, R.; Rampersaud, R.; Wu, G.W.Y.; Hough, C.M.; Epel, E.; Prather, A.A.; et al. “GrimAge,” an epigenetic predictor of mortality, is accelerated in major depressive disorder. Transl. Psychiatry 2021, 11, 193. [Google Scholar] [CrossRef]
- Yang, R.; Wu, G.W.Y.; Verhoeven, J.E.; Gautam, A.; Reus, V.I.; Kang, J.I.; Flory, J.D.; Abu-Amara, D.; Hood, L.; Doyle, F.J.; et al. A DNA methylation clock associated with age-related illnesses and mortality is accelerated in men with combat PTSD. Mol. Psychiatry 2020, 26, 4999–5009. [Google Scholar] [CrossRef]
- Horvath, S.; Levine, A.J. HIV-1 Infection Accelerates Age According to the Epigenetic Clock. J. Infect. Dis. 2015, 212, 1563–1573. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, C.; Pirazzini, C.; Sala, C.; Sambati, L.; Yusipov, I.; Kalyakulina, A.; Ravaioli, F.; Kwiatkowska, K.M.; Durso, D.F.; Ivanchenko, M.; et al. A Meta-Analysis of Brain DNA Methylation Across Sex, Age, and Alzheimer’s Disease Points for Accelerated Epigenetic Aging in Neurodegeneration. Front. Aging Neurosci. 2021, 13, 639428. [Google Scholar] [CrossRef]
- Horvath, S.; Langfelder, P.; Kwak, S.; Aaronson, J.; Rosinski, J.; Vogt, T.F.; Eszes, M.; Faull, R.L.M.; Curtis, M.A.; Waldvogel, H.J.; et al. Huntington’s disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging 2016, 8, 1485–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fingelkurts, A.A.; Fingelkurts, A.A. Operational Architectonics of the Human Brain Biopotential Field: Towards Solving the Mind-Brain Problem. Brain Mind. 2001, 2, 261–296. [Google Scholar] [CrossRef]
- Fingelkurts, A.A.; Fingelkurts, A.A.; Neves, C.F.H. Natural world physical, brain operational, and mind phenomenal space–time. Phys. Life Rev. 2010, 7, 195–249. [Google Scholar] [CrossRef] [Green Version]
- Fingelkurts, A.A.; Fingelkurts, A.A.; Neves, C.F.H. Consciousness as a phenomenon in the operational architectonics of brain organization: Criticality and self-organization considerations. Chaos Solitons Fractals 2013, 55, 13–31. [Google Scholar] [CrossRef]
- Mitina, M.; Young, S.; Zhavoronkov, A. Psychological aging, depression, and well-being. Aging 2020, 12, 18765–18777. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Kochetov, K.; Diamandis, P.; Mitina, M. PsychoAge and SubjAge: Development of deep markers of psychological and subjective age using artificial intelligence. Aging 2020, 12, 23548–23577. [Google Scholar] [CrossRef] [PubMed]
- Westerhof, G.J.; Wurm, S. Longitudinal Research on Subjective Aging, Health, and Longevity: Current Evidence and New Directions for Research. In Annual Review of Gerontology and Geriatrics; Diehl, M., Wahl, H.-W., Eds.; Subjective Aging: New Developments and Future Directions; Springer: Berlin/Heidelberg, Germany, 2015; Volume 35, pp. 145–165. [Google Scholar]
- Stephan, Y.; Sutin, A.R.; Luchetti, M.; Terracciano, A. An older subjective age is related to accelerated epigenetic aging. Psychol. Aging 2021, 36, 767–772. [Google Scholar] [CrossRef]
- Stephan, Y.; Sutin, A.R.; Terracciano, A. Subjective Age and Mortality in Three Longitudinal Samples. Psychosom. Med. 2018, 80, 659–664. [Google Scholar] [CrossRef]
- Lahav, Y.; Avidor, S.; Stein, J.Y.; Zhou, X.; Solomon, Z. Telomere Length and Depression Among Ex-Prisoners of War: The Role of Subjective Age. J. Gerontol. Ser. B 2020, 75, 21–29. [Google Scholar] [CrossRef]
- Preston, R.J. Telomeres, Telomerase and Chromosome Stability. Radiat. Res. 1997, 147, 529. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular Senescence in Aging Primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Keyes, C.L.; Westerhof, G.J. Chronological and subjective age differences in flourishing mental health and major depressive episode. Aging Ment. Health 2012, 16, 67–74. [Google Scholar] [CrossRef]
- Choi, N.G.; DiNitto, D.M. Felt age and cognitive-affective depressive symptoms in late life. Aging Ment. Health 2014, 18, 833–837. [Google Scholar] [CrossRef] [Green Version]
- Segel-Karpas, D.; Palgi, Y.; Shrira, A. The reciprocal relationship between depression and physical morbidity: The role of subjective age. Health Psychol. 2017, 36, 848–851. [Google Scholar] [CrossRef]
- Stephan, Y.; Caudroit, J.; Jaconelli, A.; Terracciano, A. Subjective Age and Cognitive Functioning: A 10-Year Prospective Study. Am. J. Geriatr. Psychiatry 2014, 22, 1180–1187. [Google Scholar] [CrossRef] [Green Version]
- Stephan, Y.; Sutin, A.R.; Terracciano, A. Subjective Age and Personality Development: A 10-Year Study. J. Personal. 2015, 83, 142–154. [Google Scholar] [CrossRef] [Green Version]
- Kwak, S.; Kim, H.; Chey, J.; Youm, Y. Feeling How Old I Am: Subjective Age Is Associated With Estimated Brain Age. Front. Aging Neurosci. 2018, 10, 168. [Google Scholar] [CrossRef]
- Elliott, M.L.; Caspi, A.; Houts, R.M.; Ambler, A.; Broadbent, J.M.; Hancox, R.J.; Harrington, H.L.; Hogan, S.; Keenan, R.; Knodt, A.; et al. Disparities in the pace of biological aging among midlife adults of the same chronological age have implications for future frailty risk and policy. Nat. Aging 2021, 1, 295–308. [Google Scholar] [CrossRef]
- Horowitz, A.M.; Villeda, S.A. Therapeutic potential of systemic brain rejuvenation strategies for neurodegenerative disease. F1000Research 2017, 6, 1291. [Google Scholar] [CrossRef] [Green Version]
- Ewing, G.; Ewing, E.; Hankey, A. Virtual scanning: A new method of health assessment and treatment. Part I: Assessment. J. Altern. Complement. Med. 2007, 13, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Besedovsky, H.O.; del Rey, A. Immune-neuro-endocrine interactions: Facts and hypotheses. Endocr. Rev. 1996, 17, 64–102. [Google Scholar] [CrossRef]
- Harbuz, M. Neuroendocrine-immune interactions. Trends Endocrinol. Metab. 2003, 14, 51–52. [Google Scholar] [CrossRef] [PubMed]
- Fingelkurts, A.A.; Fingelkurts, A.A. Brain space and time in mental disorders: Paradigm shift in biological psychiatry. Int. J. Psychiatry Med. 2019, 54, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.; Kurt, A.; Jellinger Institute of Clinical Neurobiology. Vienna; Austria Cerebral Multimorbidity in Aging. J. Neurol. Neuromed. 2016, 1, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Dow, W.H.; Philipson, T.J.; Sala-I-Martin, X. Longevity Complementarities Under Competing Risks. Am. Econ. Rev. 1999, 89, 1358–1371. [Google Scholar] [CrossRef]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD+ and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef]
- Guarente, L. Calorie restriction and sirtuins revisited. Genes Dev. 2013, 27, 2072–2085. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.-H.; Lu, M.; Lee, B.-Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef] [Green Version]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S.-I. Sirt1 Extends Life Span and Delays Aging in Mice through the Regulation of Nk2 Homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef] [Green Version]
- Mamoshina, P.; Kochetov, K.; Putin, E.; Cortese, F.; Aliper, A.; Lee, W.-S.; Ahn, S.-M.; Uhn, L.; Skjodt, N.; Kovalchuk, O.; et al. Population Specific Biomarkers of Human Aging: A Big Data Study Using South Korean, Canadian, and Eastern European Patient Populations. J. Gerontol. Ser. A 2018, 73, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Mamoshina, P.; Volosnikova, M.; Ozerov, I.V.; Putin, E.; Skibina, E.; Cortese, F.; Zhavoronkov, A. Machine Learning on Human Muscle Transcriptomic Data for Biomarker Discovery and Tissue-Specific Drug Target Identification. Front. Genet. 2018, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Galkin, F.; Mamoshina, P.; Aliper, A.; Putin, E.; Moskalev, V.; Gladyshev, V.N.; Zhavoronkov, A. Human Gut Microbiome Aging Clock Based on Taxonomic Profiling and Deep Learning. iScience 2020, 23, 101199. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115, Erratum in Genome Biol. 2015, 16, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Carmona, J.J.; Michan, S. Biology of Healthy Aging and Longevity. Rev. Investig. Clin. 2016, 68, 7–16. [Google Scholar]
- Gomez-Verjan, J.C.; Vazquez-Martinez, E.R.; Rivero-Segura, N.A.; Medina-Campos, R.H. The RNA world of human ageing. Hum. Genet. 2018, 137, 865–879. [Google Scholar] [CrossRef]
- Shireby, G.L.; Davies, J.P.; Francis, P.T.; Burrage, J.; Walker, E.M.; Neilson, G.W.A.; Dahir, A.; Thomas, A.J.; Love, S.; Smith, R.G.; et al. Recalibrating the epigenetic clock: Implications for assessing biological age in the human cortex. Brain 2020, 143, 3763–3775. [Google Scholar] [CrossRef] [PubMed]
- Lunnon, K.; Smith, R.; Hannon, E.; De Jager, P.L.; Srivastava, G.; Volta, M.; Troakes, C.; Al-Sarraj, S.; Burrage, J.; Macdonald, R.; et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat. Neurosci. 2014, 17, 1164–1170. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Chibnik, L.B.; Srivastava, G.P.; Pochet, N.; Yang, J.; Xu, J.; Kozubek, J.; Obholzer, N.; Leurgans, S.E.; Schneider, J.A.; et al. Association of brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol. 2015, 72, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M.E.; Lu, A.T.; Bennett, D.A.; Horvath, S. Epigenetic age of the prefrontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging 2015, 7, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.-U.; Bennett, D.A.; De Jager, P.L. The epigenome in Alzheimer’s disease: Current state and approaches for a new path to gene discovery and understanding disease mechanism. Acta Neuropathol. 2016, 132, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Khoury, L.Y.; Gorrie-Stone, T.; Smart, M.; Hughes, A.; Bao, Y.; Andrayas, A.; Burrage, J.; Hannon, E.; Kumari, M.; Mill, J.; et al. Systematic underestimation of the epigenetic clock and age acceleration in older subjects. Genome Biol. 2019, 20, 283. [Google Scholar] [CrossRef] [Green Version]
- Teeuw, J.; Ori, A.P.S.; Brouwer, R.M.; de Zwarte, S.M.C.; Schnack, H.G.; Hulshoff Pol, H.E.; Ophoff, R.A. Accelerated aging in the brain, epigenetic aging in blood, and polygenic risk for schizophrenia. Schizophr. Res. 2021, 231, 189–197. [Google Scholar] [CrossRef]
- Purves, D.; Augustine, G.J.; Fitzpatrick, D.; Hall, W.C.; LaMantia, A.S.; McNamara, J.O.; White, L.E. Neuroscience, 4th ed.; Sinauer Associates, Inc.: Sunderland, MA, USA, 2008. [Google Scholar]
- Fjell, A.M.; Walhovd, K.B. Structural Brain Changes in Aging: Courses, Causes and Cognitive Consequences. Rev. Neurosci. 2010, 21, 187–222. [Google Scholar] [CrossRef]
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Svennerholm, L.; Boström, K.; Jungbjer, B. Changes in weight and compositions of major membrane components of human brain during the span of adult human life of Swedes. Acta Neuropathol. 1997, 94, 345–352. [Google Scholar] [CrossRef]
- Andrews-Hanna, J.R.; Snyder, A.Z.; Vincent, J.L.; Lustig, C.; Head, D.; Raichle, M.E.; Buckner, R.L. Disruption of Large-Scale Brain Systems in Advanced Aging. Neuron 2007, 56, 924–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaie, K.W. The course of adult intellectual development. Am. Psychol. 1994, 49, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Park, D.C.; Lautenschlager, G.; Hedden, T.; Davidson, N.S.; Smith, A.D.; Smith, P.K. Models of visuospatial and verbal memory across the adult life span. Psychol. Aging 2002, 17, 299–320. [Google Scholar] [CrossRef]
- Dickie, D.A.; Job, D.E.; Gonzalez, D.R.; Shenkin, S.D.; Ahearn, T.S.; Murray, A.D.; Wardlaw, J.M. Variance in brain volume with advancing age: Implications for defining the limits of normality. PLoS ONE 2013, 8, e84093. [Google Scholar] [CrossRef] [Green Version]
- Bittner, N.; Jockwitz, C.; Franke, K.; Gaser, C.; Moebus, S.; Bayen, U.J.; Amunts, K.; Caspers, S. When your brain looks older than expected: Combined lifestyle risk and BrainAGE. Anat. Embryol. 2021, 226, 621–645. [Google Scholar] [CrossRef]
- Jagust, W. Vulnerable Neural Systems and the Borderland of Brain Aging and Neurodegeneration. Neuron 2013, 77, 219–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efranke, K.; Eristow, M.; Egaser, C. Gender-specific impact of personal health parameters on individual brain aging in cognitively unimpaired elderly subjects. Front. Aging Neurosci. 2014, 6, 94. [Google Scholar] [CrossRef] [Green Version]
- Eavani, H.; Habes, M.; Satterthwaite, T.D.; An, Y.; Hsieh, M.-K.; Honnorat, N.; Erus, G.; Doshi, J.; Ferrucci, L.; Beason-Held, L.L.; et al. Heterogeneity of structural and functional imaging patterns of advanced brain aging revealed via machine learning methods. Neurobiol. Aging 2018, 71, 41–50. [Google Scholar] [CrossRef]
- Bartrés-Faz, D.; Arenaza-Urquijo, E.M. Structural and Functional Imaging Correlates of Cognitive and Brain Reserve Hypotheses in Healthy and Pathological Aging. Brain Topogr. 2011, 24, 340–357. [Google Scholar] [CrossRef]
- Ziegler, G.; Dahnke, R.; Gaser, C.; Alzheimer’s Disease Neuroimaging Initiative. Models of the aging brain structure and individual decline. Front. Neuroinform. 2012, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Sperling, R.; Mormino, E.; Johnson, K. The Evolution of Preclinical Alzheimer’s Disease: Implications for Prevention Trials. Neuron 2014, 84, 608–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, M.L. MRI-Based biomarkers of accelerated aging and dementia risk in midlife: How close are we? Ageing Res. Rev. 2020, 61, 101075. [Google Scholar] [CrossRef] [PubMed]
- Franke, K.; Ziegler, G.; Klöppel, S.; Gaser, C.; Alzheimer’s Disease Neuroimaging Initiative. Estimating the age of healthy subjects from T1-weighted MRI scans using kernel methods: Exploring the influence of various parameters. NeuroImage 2010, 50, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.H.; Franke, K. Predicting Age Using Neuroimaging: Innovative Brain Ageing Biomarkers. Trends Neurosci. 2017, 40, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.H.; Poudel, R.P.; Tsagkrasoulis, D.; Caan, M.W.; Steves, C.; Spector, T.D.; Montana, G. Predicting brain age with deep learning from raw imaging data results in a reliable and heritable biomarker. NeuroImage 2017, 163, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Liem, F.; Varoquaux, G.; Kynast, J.; Beyer, F.; Masouleh, S.K.; Huntenburg, J.M.; Lampe, L.; Rahim, M.; Abraham, A.; Craddock, R.C.; et al. Predicting brain-age from multimodal imaging data captures cognitive impairment. Neuroimage 2017, 148, 179–188. [Google Scholar] [CrossRef]
- Enzinger, C.; Fazekas, F.; Matthews, P.M.; Ropele, S.; Schmidt, H.; Smith, S.; Schmidt, R. Risk factors for progression of brain atrophy in aging: Six-year follow-up of normal subjects. Neurology 2005, 64, 1704–1711. [Google Scholar] [CrossRef]
- Franke, K.; Gaser, C.; Manor, B.; Novak, V. Advanced BrainAGE in older adults with type 2 diabetes mellitus. Front. Ageing Neurosci. 2013, 5, 90. [Google Scholar]
- Fingelkurts, A.A.; Fingelkurts, A.A.; Bagnato, S.; Boccagni, C.; Galardi, G. Dissociation of vegetative and minimally conscious patients based on brain Operational Architectonics: Factor of etiology. Clin. EEG Neurosci. 2013, 44, 209–220. [Google Scholar] [CrossRef]
- Rudrauf, D. Structure-Function Relationships behind the Phenomenon of Cognitive Resilience in Neurology: Insights for Neuroscience and Medicine. Adv. Neurosci. 2014, 2014, 462765. [Google Scholar] [CrossRef] [Green Version]
- Luria, A. The Man with a Shattered World: The History of a Brain Wound; Harvard University Press: Cambridge, MA, USA, 1987. [Google Scholar]
- Lee, R.G.; van Donkelaar, P. Mechanisms underlying functional recovery following stroke. Can. J. Neurol. Sci. 1995, 22, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noppeney, U.; Friston, K.J.; Price, C.J. Degenerate neuronal systems sustaining cognitive functions. J. Anat. 2004, 205, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Philippi, C.L.; Feinstein, J.S.; Khalsa, S.S.; Damasio, A.; Tranel, D.; Landini, G.; Williford, K.; Rudrauf, D. Preserved Self-Awareness following Extensive Bilateral Brain Damage to the Insula, Anterior Cingulate, and Medial Prefrontal Cortices. PLoS ONE 2012, 7, e38413. [Google Scholar] [CrossRef] [Green Version]
- Bagnato, S.; Boccagni, C.; Sant’Angelo, A.; Fingelkurts, A.A.; Fingelkurts, A.A.; Galardi, G. Emerging from an unresponsive wakefulness syndrome: Brain plasticity has to cross a threshold level. Neurosci. Biobehav. Rev. 2013, 37, 2721–2736. [Google Scholar] [CrossRef] [PubMed]
- Fingelkurts, A.A.; Fingelkurts, A.A. Longitudinal dynamics of 3-dimensional components of selfhood after severe traumatic brain injury: A qEEG case study. Clin. EEG Neurosci. 2017, 48, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Price, J.L.; Morris, J.C. Tangles and plaques in nondemented ageing and “preclinical” Alzheimer’s disease. Ann. Neurol. 1999, 45, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Cummings, J.L.; DeKosky, S.T.; Barberger-Gateau, P.; Delacourte, A.; Frisoni, G.; Fox, N.C.; Galasko, D.; et al. Revising the definition of Alzheimer’s disease: A new lexicon. Lancet Neurol. 2010, 9, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Penfield, W. Neurophysiological Basis of the Higher Functions of the Nervous System—Introduction. In Handbook of Physiology. A Critical, Comprehensive Presentation of Physiological Knowledge and Concepts; Field, J., Ed.; American Physiological Society: Washington, DC, USA, 1960; pp. 1441–1445. [Google Scholar]
- Lopes da Silva, F.H. Neural mechanisms underlying brain waves: From neural membranes to networks. Electroencephalogr. Clin. Neurophysiol 1991, 79, 81–93. [Google Scholar] [CrossRef]
- Nunez, P.L.; Williamson, S.J. Neocortical Dynamics and Human EEG Rhythms; Oxford University Press: New York, NY, USA, 1995. [Google Scholar]
- Nuwer, M. Assessment of digital EEG, quantitative EEG, and EEG brain mapping: Report of the American Academy of Neurology and the American Clinical Neurophysiology Society. Neurology 1997, 49, 277–292. [Google Scholar] [CrossRef] [Green Version]
- Gasser, T.; Bächer, P.; Steinberg, H. Test-retest reliability of spectral parameters of the EEG. Electroencephalogr. Clin. Neurophysiol. 1985, 60, 312–319. [Google Scholar] [CrossRef]
- Stassen, H.H.; Bomben, G.; Propping, P. Genetic aspects of the EEG: An investigation into the within-pair similarity of monozigotic and dyzigotic twins with a new method of analysis. Electroencephalogr. Clin. Neurophysiol. 1987, 66, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Stassen, H.H.; Bomben, G.; Hell, D. Familial brain wave patterns: Study of a 12-sib family. Psychiatr. Genet. 1998, 8, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Pollock, V.; Schneider, L.; Lyness, S. Reliability of topographic quantitative EEG amplitude in healthy late-middle-aged and elderly subjects. Electroencephalogr. Clin. Neurophysiol. 1991, 79, 20–26. [Google Scholar] [CrossRef]
- Fingelkurts, A.A.; Fingelkurts, A.A.; Ermolaev, V.A.; Kaplan, A.Y. Stability, reliability and consistency of the compositions of brain oscillations. Int. J. Psychophysiol. 2006, 59, 116–126. [Google Scholar] [CrossRef]
- Posthuma, D.; Neale, M.C.; Boomsma, D.I.; de Geus, E.J.C. Are Smarter Brains Running Faster? Heritability of Alpha Peak Frequency, IQ, and Their Interrelation. Behav. Genet. 2001, 31, 567–579. [Google Scholar] [CrossRef]
- van Beijsterveldt, C.; van Baal, G. Twin and family studies of the human electroencephalogram: A review and a meta-analysis. Biol. Psychol. 2002, 61, 111–138. [Google Scholar] [CrossRef] [PubMed]
- Anokhin, A.P.; Müller, V.; Lindenberger, U.; Heath, A.C.; Myers, E. Genetic influences on dynamic complexity of brain oscillations. Neurosci. Lett. 2006, 397, 93–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyberg, L.; Pudas, S. Successful Memory Aging. Annu. Rev. Psychol. 2019, 70, 219–243. [Google Scholar] [CrossRef]
- da Silva, F.L.; Vos, J.; Mooibroek, J.; van Rotterdam, A. Relative contributions of intracortical and thalamo-cortical processes in the generation of alpha rhythms, revealed by partial coherence analysis. Electroencephalogr. Clin. Neurophysiol. 1980, 50, 449–456. [Google Scholar] [CrossRef]
- Bhattacharya, B.S.; Coyle, D.; Maguire, L.P. A thalamo-cortico-thalamic neural mass model to study alpha rhythms in Alzheimer’s disease. Neural Netw. 2011, 24, 631–645. [Google Scholar] [CrossRef]
- Garcés, P.; Vicente, R.; Wibral, M.; Pineda-Pardo, J.; López, M.E.; Aurtenetxe, S.; Marcos, A.; de Andrés, M.E.; Yus, M.; Sancho, M.; et al. Brain-wide slowing of spontaneous alpha rhythms in mild cognitive impairment. Front. Aging Neurosci. 2013, 5, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimesch, W. EEG alpha and theta oscillations reflect cognitive and memory performance: A review and analysis. Brain Res. Rev. 1999, 29, 169–195. [Google Scholar] [CrossRef] [PubMed]
- Bressler, S.L.; Kelso, J. Cortical coordination dynamics and cognition. Trends Cogn. Sci. 2001, 5, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Fingelkurts, A.A.; Fingelkurts, A.A. Short-term EEG spectral pattern as a single event in EEG phenomenology. Open Neuroimaging J. 2010, 4, 130–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Başar, E. A review of alpha activity in integrative brain function: Fundamental physiology, sensory coding, cognition and pathology. Int. J. Psychophysiol. 2012, 86, 1–24. [Google Scholar] [CrossRef]
- Matejcek, M. Some relationships between occipital E.E.G. activity and age. A spectral analytic study. Rev. Electroencephalogr. Neurophysiol. Clin. 1980, 10, 122–130. [Google Scholar] [CrossRef]
- Köpruner, V.; Pfurtscheller, G.; Auer, L. Quantitative EEG in Normals and in Patients with Cerebral Ischemia. Prog. Brain Res. 1984, 62, 29–50. [Google Scholar] [CrossRef]
- Babiloni, C.; Binetti, G.; Cassarino, A.; Forno, G.D.; Del Percio, C.; Ferreri, F.; Ferri, R.; Frisoni, G.B.; Galderisi, S.; Hirata, K.; et al. Sources of cortical rhythms in adults during physiological aging: A multicentric EEG study. Hum. Brain Mapp. 2006, 27, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Knyazeva, M.G.; Barzegaran, E.; Vildavski, V.Y.; Demonet, J.-F. Aging of human alpha rhythm. Neurobiol. Aging 2018, 69, 261–273. [Google Scholar] [CrossRef]
- Purdon, P.L.; Pavone, K.J.; Akeju, O.; Smith, A.C.; Sampson, A.L.; Lee, J.; Zhou, D.W.; Solt, K.; Brown, E.N. The Ageing Brain: Age-dependent changes in the electroencephalogram during propofol and sevoflurane general anaesthesia. Br. J. Anaesth. 2015, 115 (Suppl. S1), i46–i57. [Google Scholar] [CrossRef] [Green Version]
- Dukart, J.; Schroeter, M.L.; Mueller, K.; Alzheimer’s Disease Neuroimaging Initiative. Age Correction in Dementia—Matching to a Healthy Brain. PLoS ONE 2011, 6, e22193. [Google Scholar] [CrossRef] [PubMed]
- Szelies, B.; Mielke, R.; Kessler, J.; Heiss, W.D. EEG power changes are related with regional cerebral glucose metbolism in vascular dementia. Clin. Neurophysiol. 1999, 110, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Leocani, L.; Locatelli, T.; Martinelli, V.; Rovaris, M.; Falautano, M.; Filippi, M.; Magnani, G.; Comi, G. Electroencephalographic coherence analysis in multiple sclerosis: Correlation with clinical, neuropsychological, and MRI findings. J. Neurol. Neurosurg. Psychiatry 2000, 69, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, A.; Hornero, R.; Mayo, A.; Poza, J.; Gil-Gregorio, P.; Ortiz, T. EEG spectral profile in Alzheimer’s disease and mild cognitive impairment. Clin. Neurophysiol. 2006, 117, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Medaglia, J.D.; Pasqualetti, F.; Hamilton, R.H.; Thompson-Schill, S.L.; Bassett, D.S. Brain and cognitive reserve: Translation via network control theory. Neurosci. Biobehav. Rev. 2017, 75, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Stern, Y. Cognitive Reserve and Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2006, 20, 112–117. [Google Scholar] [CrossRef]
- Cespón, J.; Miniussi, C.; Pellicciari, M.C. Interventional programmes to improve cognition during healthy and pathological ageing: Cortical modulations and evidence for brain plasticity. Ageing Res. Rev. 2018, 43, 81–98. [Google Scholar] [CrossRef]
- Lã3Pez, M.E.; Aurtenetxe, S.; Pereda, E.; Cuesta, P.; Castellanos, N.; Bruã±A, R.; Niso, G.; Maestu, F.; Bajo, R. Cognitive reserve is associated with the functional organization of the brain in healthy aging: A MEG study. Front. Aging Neurosci. 2014, 6, 125. [Google Scholar] [CrossRef]
- Zarahn, E.; Rakitin, B.; Abela, D.; Flynn, J.; Stern, Y. Age-related changes in brain activation during a delayed item recognition task. Neurobiol. Aging 2007, 28, 784–798. [Google Scholar] [CrossRef]
- Steffener, J.; Stern, Y. Exploring the neural basis of cognitive reserve in aging. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 467–473. [Google Scholar] [CrossRef] [Green Version]
- Stern, Y.; Habeck, C.; Moeller, J.; Scarmeas, N.; Anderson, K.E.; Hilton, H.J.; Flynn, J.; Sackeim, H.; van Heertum, R. Brain Networks Associated with Cognitive Reserve in Healthy Young and Old Adults. Cereb. Cortex 2005, 15, 394–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Zoubi, O.; Wong, C.K.; Kuplicki, R.T.; Yeh, H.-W.; Mayeli, A.; Refai, H.; Paulus, M.; Bodurka, J. Predicting Age From Brain EEG Signals—A Machine Learning Approach. Front. Aging Neurosci. 2018, 10, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Paixao, L.; Oliva, J.T.; Goparaju, B.; Carvalho, D.; van Leeuwen, K.G.; Akeju, O.; Thomas, R.J.; Cash, S.S.; Bianchi, M.T.; et al. Brain age from the electroencephalogram of sleep. Neurobiol. Aging 2019, 74, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Gontier, G.; Iyer, M.; Shea, J.M.; Bieri, G.; Wheatley, E.G.; Ramalho-Santos, M.; Villeda, S.A. Tet2 Rescues Age-Related Regenerative Decline and Enhances Cognitive Function in the Adult Mouse Brain. Cell Rep. 2018, 22, 1974–1981. [Google Scholar] [CrossRef] [Green Version]
- Fahy, G.M.; Brooke, R.T.; Watson, J.P.; Good, Z.; Vasanawala, S.S.; Maecker, H.; Leipold, M.D.; Lin, D.T.S.; Kobor, M.S.; Horvath, S. Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell 2019, 18, e13028. [Google Scholar] [CrossRef] [Green Version]
- Katsimpardi, L.; Litterman, N.K.; Schein, P.A.; Miller, C.M.; Loffredo, F.S.; Wojtkiewicz, G.R.; Chen, J.W.; Lee, R.T.; Wagers, A.J.; Rubin, L.L. Vascular and Neurogenic Rejuvenation of the Aging Mouse Brain by Young Systemic Factors. Science 2014, 344, 630–634. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, M.; Saarma, M.; Lindholm, P. Unconventional neurotrophic factors CDNF and MANF: Structure, physiological functions and therapeutic potential. Neurobiol. Dis. 2017, 97, 90–102. [Google Scholar] [CrossRef]
- White, C.W.; Pratt, K.; Villeda, S.A. OPCs on a Diet: A Youthful Serving of Remyelination. Cell Metab. 2019, 30, 1004–1006. [Google Scholar] [CrossRef]
- Horowitz, A.M.; Fan, X.; Bieri, G.; Smith, L.K.; Sanchez-Diaz, C.I.; Schroer, A.B.; Gontier, G.; Casaletto, K.B.; Kramer, J.H.; Williams, K.E.; et al. Blood factors transfer beneficial effects of exercise on neurogenesis and cognition to the aged brain. Science 2020, 369, 167–173. [Google Scholar] [CrossRef]
- Navarro, A.; Gómez, C.G.; López-Cepero, J.M.; Boveris, A. Beneficial effects of moderate exercise on mice aging: Survival, behavior, oxidative stress, and mitochondrial electron transfer. Am. J. Physiol. Integr. Comp. Physiol. 2004, 286, R505–R511. [Google Scholar] [CrossRef] [Green Version]
- Melov, S.; Tarnopolsky, M.A.; Beckman, K.; Felkey, K.; Hubbard, A. Resistance Exercise Reverses Aging in Human Skeletal Muscle. PLoS ONE 2007, 2, e465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poljsak, B.; Milisav, I. NAD+ as the Link Between Oxidative Stress, Inflammation, Caloric Restriction, Exercise, DNA Repair, Longevity, and Health Span. Rejuvenation Res. 2016, 19, 406–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quach, A.; Levine, M.E.; Tanaka, T.; Lu, A.T.; Chen, B.H.; Ferrucci, L.; Ritz, B.; Bandinelli, S.; Neuhouser, M.L.; Beasley, J.M.; et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging 2017, 9, 419–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoro, E.J. Overview of caloric restriction and ageing. Mech. Ageing Dev. 2005, 126, 913–922. [Google Scholar] [CrossRef]
- Vera, E.; de Jesus, B.B.; Foronda, M.; Flores, J.M.; Blasco, M.A. Telomerase Reverse Transcriptase Synergizes with Calorie Restriction to Increase Health Span and Extend Mouse Longevity. PLoS ONE 2013, 8, e53760. [Google Scholar] [CrossRef] [Green Version]
- Waterland, R.A.; Jirtle, R.L. Transposable Elements: Targets for Early Nutritional Effects on Epigenetic Gene Regulation. Mol. Cell. Biol. 2003, 23, 5293–5300. [Google Scholar] [CrossRef] [Green Version]
- Hore, T.A. Modulating epigenetic memory through vitamins and TET: Implications for regenerative medicine and cancer treatment. Epigenomics 2017, 9, 863–871. [Google Scholar] [CrossRef]
- Alehagen, U.; Aaseth, J.; Alexander, J.; Johansson, P. Still reduced cardiovascular mortality 12 years after supplementation with selenium and coenzyme Q10 for four years: A validation of previous 10-year follow-up results of a prospective randomized double-blind placebo-controlled trial in elderly. PLoS ONE 2018, 13, e0193120. [Google Scholar] [CrossRef] [Green Version]
- Comhaire, F.; Decleer, W. Can the biological mechanisms of ageing be corrected by food supplementation. The concept of health care over sick care. Aging Male 2020, 23, 1146–1157. [Google Scholar] [CrossRef]
- Proshkina, E.; Shaposhnikov, M.; Moskalev, A. Genome-Protecting Compounds as Potential Geroprotectors. Int. J. Mol. Sci. 2020, 21, 4484. [Google Scholar] [CrossRef]
- Nur, S.M.; Rath, S.; Ahmad, V.; Ahmad, A.; Ateeq, B.; Khan, M.I. Nutritive vitamins as epidrugs. Crit. Rev. Food Sci. Nutr. 2021, 61, 1712674. [Google Scholar] [CrossRef] [PubMed]
- Stanfel, M.N.; Shamieh, L.S.; Kaeberlein, M.; Kennedy, B.K. The TOR pathway comes of age. Biochim. Biophys. Acta 2009, 1790, 1067–1074. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, B.; Lamming, D.W. The Mechanistic Target of Rapamycin: The Grand ConducTOR of Metabolism and Aging. Cell Metab. 2016, 23, 990–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976, Erratum in Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shea, T.B.; Remington, R. Nutritional supplementation for Alzheimer’s disease? Curr. Opin. Psychiatry 2015, 28, 141–147. [Google Scholar] [CrossRef]
- Dominguez, L.J.; Barbagallo, M. Nutritional prevention of cognitive decline and dementia. Acta Biomed. 2018, 89, 276–290. [Google Scholar]
- Corpas, R.; Griñán-Ferré, C.; Rodriguez-Farre, E.; Pallàs, M.; Sanfeliu, C. Resveratrol Induces Brain Resilience Against Alzheimer Neurodegeneration Through Proteostasis Enhancement. Mol. Neurobiol. 2019, 56, 1502–1516. [Google Scholar] [CrossRef] [Green Version]
- Orozco-Solis, R.; Ramadori, G.; Coppari, R.; Sassone-Corsi, P. SIRT1 Relays Nutritional Inputs to the Circadian Clock Through the Sf1 Neurons of the Ventromedial Hypothalamus. Endocrinology 2015, 156, 2174–2184. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.-I.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Verdin, E. The Many Faces of Sirtuins: Coupling of NAD metabolism, sirtuins and lifespan. Nat. Med. 2014, 20, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Chastin, S.F.M.; Dall, P.; Tigbe, W.; Grant, P.M.; Ryan, C.; Rafferty, D.; Granat, M. Compliance with physical activity guidelines in a group of UK-based postal workers using an objective monitoring technique. Eur. J. Appl. Physiol. 2009, 106, 893–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theofilou, P.; Saborit, A.R. Adherence and physical activity. Health Psychol. Res. 2013, 1, e6. [Google Scholar] [CrossRef]
- Gomes, T.N.; Katzmarzyk, P.T.; Hedeker, D.; Fogelholm, M.; Standage, M.; Onywera, V.; Lambert, E.V.; Tremblay, M.S.; Chaput, J.-P.; Tudor-Locke, C.; et al. Correlates of compliance with recommended levels of physical activity in children. Sci. Rep. 2017, 7, 16507. [Google Scholar] [CrossRef] [Green Version]
- Heilbronn, L.K.; de Jonge, L.; Frisard, M.I.; DeLany, J.P.; Larson-Meyer, D.E.; Rood, J.; Nguyen, T.; Martin, C.K.; Volaufova, J.; Most, M.M.; et al. Effect of 6-month calorie restriction on biomarkers of longevity, metabolic adaptation, and oxidative stress in overweight individuals: A randomized controlled trial. JAMA 2006, 295, 1539–1548. [Google Scholar] [CrossRef]
- Ravussin, E.; Redman, L.M.; Rochon, J.; Das, S.K.; Fontana, L.; Kraus, W.E.; Romashkan, S.; Williamson, D.A.; Meydani, S.N.; Villareal, D.T.; et al. A 2-Year Randomized Controlled Trial of Human Caloric Restriction: Feasibility and Effects on Predictors of Health Span and Longevity. J. Gerontol. Ser. A 2015, 70, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Romashkan, S.V.; Das, S.K.; Villareal, D.T.; Ravussin, E.; Redman, L.M.; Rochon, J.; Bhapkar, M.; Kraus, W.E.; CALERIE Study Group. Safety of two-year caloric restriction in non-obese healthy individuals. Oncotarget 2016, 7, 19124–19133. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, S.G.; Boyd, P.; Bailey, C.P.; Shams-White, M.M.; Agurs-Collins, T.; Hall, K.; Reedy, J.; Sauter, E.R.; Czajkowski, S.M. Perspective: Time-Restricted Eating Compared with Caloric Restriction: Potential Facilitators and Barriers of Long-Term Weight Loss Maintenance. Adv. Nutr. Int. Rev. J. 2021, 12, 325–333. [Google Scholar] [CrossRef]
- Madeo, F.; Carmona-Gutierrez, D.; Hofer, S.J.; Kroemer, G. Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab. 2019, 29, 592–610. [Google Scholar] [CrossRef] [Green Version]
- Guerrieri, D.; Moon, H.Y.; van Praag, H. Exercise in a Pill: The Latest on Exercise-Mimetics. Brain Plast. 2017, 2, 153–169. [Google Scholar] [CrossRef] [Green Version]
- Fingelkurts, A.A.; Fingelkurts, A.A.; Neves, C.F.H. Neuro-assessment of leadership training. Coaching Int. J. Theory Res. Pract. 2020, 13, 107–145. [Google Scholar] [CrossRef]
- Wilson, K.A.; Chamoli, M.; Hilsabeck, T.A.; Pandey, M.; Bansal, S.; Chawla, G.; Kapahi, P. Evaluating the beneficial effects of dietary restrictions: A framework for precision nutrigeroscience. Cell Metab. 2021, 33, 2142–2173. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Stout, M.B.; Sierra, F. Resilience in Aging Mice. J. Gerontol. Ser. A 2016, 71, 1407–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.F.; Seals, D.R.; Hamilton, K.L. A viewpoint on considering physiological principles to study stress resistance and resilience with aging. Ageing Res. Rev. 2017, 38, 1–5. [Google Scholar] [CrossRef] [PubMed]
- DE Grey, A.D.N.J.; Ames, B.N.; Andersen, J.K.; Bartke, A.; Campisi, J.; Heward, C.B.; McCARTER, R.J.M.; Stock, G. Time to Talk SENS: Critiquing the Immutability of Human Aging. Ann. N. Y. Acad. Sci. 2002, 959, 452–462. [Google Scholar] [CrossRef]
- Fingelkurts, A.A.; Fingelkurts, A.A. Operational Architectonics methodology for EEG analysis: Theory and results. Neuromethods 2015, 91, 1–59. [Google Scholar]
- Kurgansky, A.V. Functional Organization of the Human Brain in the Resting State. Neurosci. Behav. Physiol. 2019, 49, 1135–1144. [Google Scholar] [CrossRef]
- Buzsáki, G. Rhythms of the Brain; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- Lazarev, V.V. The relationship of theory and methodology in EEG studies of mental activity. Int. J. Psychophysiol. 2006, 62, 384–393. [Google Scholar] [CrossRef]
- Wacker, J.; Chavanon, M.-L.; Stemmler, G. Resting EEG signatures of agentic extraversion: New results and meta-analytic integration. J. Res. Pers. 2010, 44, 167–179. [Google Scholar] [CrossRef]
- Rogala, J.; Kublik, E.; Krauz, R.; Wróbel, A. Resting-state EEG activity predicts frontoparietal network reconfiguration and improved attentional performance. Sci. Rep. 2020, 10, 5064. [Google Scholar] [CrossRef] [Green Version]
- Fingelkurts, A.A.; Fingelkurts, A.A.; Kallio-Tamminen, T. Selfhood triumvirate: From phenomenology to brain activity and back again. Conscious. Cogn. 2020, 86, 103031. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Grossman, R.I.; Babb, J.S.; Rabin, M.L.; Mannon, L.J.; Kolson, D.L. Age-Related Total Gray Matter and White Matter Changes in Normal Adult Brain. Part I: Volumetric MR Imaging Analysis. Am. J. Neuroradiol. 2002, 23, 1327–1333. [Google Scholar]
- Raz, N.; Lindenberger, U.; Rodrigue, K.M.; Kennedy, K.M.; Head, D.; Williamson, A.; Dahle, C.; Gerstorf, D.; Acker, J.D. Regional Brain Changes in Aging Healthy Adults: General Trends, Individual Differences and Modifiers. Cereb. Cortex 2005, 15, 1676–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salthouse, T.A. When does age-related cognitive decline begin? Neurobiol. Aging 2009, 30, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh-Manoux, A.; Kivimaki, M.; Glymour, M.M.; Elbaz, A.; Berr, C.; Ebmeier, K.; Ferrie, J.E.; Dugravot, A. Timing of onset of cognitive decline: Results from Whitehall II prospective cohort study. BMJ 2012, 344, d7622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scraggs, T.L. EEG maturation: Viability through adolescence. Neurodiagnostic J. 2012, 52, 176–203. [Google Scholar]
- Kaminska, A.; Eisermann, M.; Plouin, P. Child EEG (and Maturation). In Handbook of Clinical Neurology, 3rd ed.; Levin, K.H., Chauvel, P., Eds.; Clinical Neurophysiology: Basis and Technical Aspects; Elsevier B.V.: Amsterdam, The Netherlands, 2019; Volume 160, pp. 125–142. [Google Scholar]
- Giedd, J.N.; Blumenthal, J.; Jeffries, N.O.; Castellanos, F.; Liu, H.; Zijdenbos, A.; Paus, T.; Evans, A.C.; Rapoport, J.L. Brain development during childhood and adolescence: A longitudinal MRI study. Nat. Neurosci. 1999, 2, 861–863. [Google Scholar] [CrossRef]
- Sobel, H. When Does Human Aging Start? Gerontologist 1966, 6, 17–22. [Google Scholar] [CrossRef]
- Gordon, E.; Cooper, N.; Rennie, C.; Hermens, D.; Williams, L. Integrative Neuroscience: The Role of a Standardized Database. Clin. EEG Neurosci. 2005, 36, 64–75. [Google Scholar] [CrossRef]
- Prichep, L.S. Use of Normative Databases and Statistical Methods in Demonstrating Clinical Utility of QEEG: Importance and Cautions. Clin. EEG Neurosci. 2005, 36, 82–87. [Google Scholar] [CrossRef]
- Thatcher, R.W.; Lubar, J.F. History of the Scientific Standards of QEEG Normative Databases. In Introduction to QEEG and Neurofeedback: Advanced Theory and Applications; Budzinsky, T., Budzinski, H., Evans, J., Abarbanel, A., Eds.; Academic Press: San Diego, CA, USA, 2008; pp. 29–62. [Google Scholar]
- Beck, A.T.; Ward, C.H.; Mendelson, M.; Mock, J.; Erbaugh, J. An inventory for measuring depression. Arch. Gen. Psychiatry 1961, 4, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.T.; Epstein, N.; Brown, G.; Steer, R.A. An inventory for measuring clinical anxiety: Psychometric properties. J. Consult. Clin. Psychol. 1988, 56, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M. The Assessment of Anxiety States by Rating. Psychol. Psychother. Theory Res. Pract. 1959, 32, 50–55. [Google Scholar] [CrossRef] [PubMed]
- John, O.P.; Donahue, E.M.; Kentle, R.L. The Big Five Inventory-Versions 4a and 54; University of California, Institute of Personality and Social Research: Berkeley, CA, USA, 1991. [Google Scholar]
- Potter, G.D.M.; Cade, J.; Grant, P.J.; Hardie, L.J. Nutrition and the circadian system. Br. J. Nutr. 2016, 116, 434–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suga, H.; Asakura, K.; Sasaki, S.; Nojima, M.; Okubo, H.; Hirota, N.; Notsu, A.; Fukui, M.; Date, C. Effect of seasonality on the estimated mean value of nutrients and ranking ability of a self-administered diet history questionnaire. Nutr. J. 2014, 13, 51. [Google Scholar] [CrossRef] [Green Version]
- Semeraro, M.D.; Smith, C.; Kaiser, M.; Levinger, I.; Duque, G.; Gruber, H.-J.; Herrmann, M. Physical activity, a modulator of aging through effects on telomere biology. Aging 2020, 12, 13803–13823. [Google Scholar] [CrossRef]
- Perneger, T.V. What’s wrong with Bonferroni adjustments. BMJ 1998, 316, 1236–1238. [Google Scholar] [CrossRef]
- Bland, M. An Introduction to Medical Statistics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Pavanello, S.; Campisi, M.; Fabozzo, A.; Cibin, G.; Tarzia, V.; Toscano, G.; Gerosa, G. The biological age of the heart is consistently younger than chronological age. Sci. Rep. 2020, 10, 10752. [Google Scholar] [CrossRef]
- Kioussis, B.; Tuttle, C.S.; Heard, D.S.; Kennedy, B.K.; Lautenschlager, N.T.; Maier, A.B. Targeting impaired nutrient sensing with repurposed therapeutics to prevent or treat age-related cognitive decline and dementia: A systematic review. Ageing Res. Rev. 2021, 67, 101302. [Google Scholar] [CrossRef]
- Amen, D.G.; Taylor, D.V.; Ojala, K.; Kaur, J.; Willeumier, K. Effects of brain-directed nutrients on cerebral blood flow and neuropsychological testing: A randomized, double-blind, placebo-controlled, crossover trial. Adv. Mind. Body Med. 2013, 27, 24–33. [Google Scholar]
- Morelli, M.B.; Gambardella, J.; Castellanos, V.; Trimarco, V.; Santulli, G. Vitamin C and Cardiovascular Disease: An Update. Antioxidants 2020, 9, 1227. [Google Scholar] [CrossRef]
- Young, J.I.; Züchner, S.; Wang, G. Regulation of the Epigenome by Vitamin C. Annu. Rev. Nutr. 2015, 35, 545–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, T.L.; Ahearn, E.L.; Cimmino, L. Reprogramming the Epigenome With Vitamin C. Front. Cell Dev. Biol. 2019, 7, 128. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Heintz, N. The Nuclear DNA Base 5-Hydroxymethylcytosine Is Present in Purkinje Neurons and the Brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef] [Green Version]
- Kaas, G.A.; Zhong, C.; Eason, D.E.; Ross, D.L.; Vachhani, R.V.; Ming, G.-L.; King, J.R.; Song, H.; Sweatt, J.D. TET1 Controls CNS 5-Methylcytosine Hydroxylation, Active DNA Demethylation, Gene Transcription, and Memory Formation. Neuron 2013, 79, 1086–1093. [Google Scholar] [CrossRef] [Green Version]
- Rudenko, A.; Dawlaty, M.M.; Seo, J.; Cheng, A.W.; Meng, J.; Le, T.; Faull, K.F.; Jaenisch, R.; Tsai, L.-H. Tet1 Is Critical for Neuronal Activity-Regulated Gene Expression and Memory Extinction. Neuron 2013, 79, 1109–1122. [Google Scholar] [CrossRef] [Green Version]
- Ming, G.-L.; Song, H. Adult Neurogenesis in the Mammalian Brain: Significant Answers and Significant Questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [Green Version]
- Erickson, K.I.; Voss, M.W.; Prakash, R.S.; Basak, C.; Szabo, A.; Chaddock, L.; Kim, J.S.; Heo, S.; Alves, H.; White, S.M.; et al. Exercise training increases size of hippocampus and improves memory. Proc. Natl. Acad. Sci. USA 2011, 108, 3017–3022. [Google Scholar] [CrossRef] [Green Version]
- Minhas, P.S.; Latif-Hernandez, A.; McReynolds, M.R.; Durairaj, A.S.; Wang, Q.; Rubin, A.; Joshi, A.U.; He, J.Q.; Gauba, E.; Liu, L.; et al. Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature 2021, 590, 122–128. [Google Scholar] [CrossRef]
- Etgen, T.; Sander, D.; Bickel, H.; Sander, K.; Förstl, H. Vitamin D Deficiency, Cognitive Impairment and Dementia: A Systematic Review and Meta-Analysis. Dement. Geriatr. Cogn. Disord. 2012, 33, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Haussler, M.R.; Whitfield, G.K.; Haussler, C.A.; Sabir, M.S.; Khan, Z.; Sandoval, R.; Jurutka, P.W. 1,25-dihydroxyvitamin d and klotho: A tale of two renal hormones coming of age. Vitam. Horm. 2016, 100, 165–230. [Google Scholar]
- Kuro-O, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.C.; Huang, S.M.; Chen, A.; Sun, C.Y.; Lin, S.H.; Chen, J.S.; Liu, S.T.; Hsu, Y.J. Resveratrol increases anti-aging Klotho gene expression via the activating transcription factor 3/c-Jun complex-mediated signaling pathway. Int. J. Biochem. Cell Biol. 2014, 53, 361–371. [Google Scholar] [CrossRef]
- Papadopoli, D.; Boulay, K.; Kazak, L.; Pollak, M.; Mallette, F.A.; Topisirovic, I.; Hulea, L. mTOR as a central regulator of lifespan and aging. F1000Research 2019, 8, 998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaeberlein, M. Rapamycin and Ageing: When, for How Long, and How Much? J. Genet. Genom. 2014, 41, 459–463. [Google Scholar] [CrossRef] [Green Version]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular Interplay between Mammalian Target of Rapamycin (mTOR), Amyloid-β, and Tau: Effects on Cognitive Impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Alafuzoff, I.; Soininen, H.; Winblad, B.; Pei, J.J. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005, 272, 4211–4220. [Google Scholar] [CrossRef]
- Tramutola, A.; Triplett, J.C.; Di Domenico, F.; Niedowicz, D.M.; Murphy, M.P.; Coccia, R.; Perluigi, M.; Butterfield, D.A. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): Analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J. Neurochem. 2015, 133, 739–749. [Google Scholar] [CrossRef]
- Lisse, T.S.; Hewison, M. Vitamin D: A new player in the world of mTOR signalling. Cell Cycle 2011, 10, 1888–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.C.; Patchva, S.; Aggarwal, B.B. Therapeutic Roles of Curcumin: Lessons Learned from Clinical Trials. AAPS J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Aller, G.S.; Carson, J.D.; Tang, W.; Peng, H.; Zhao, L.; Copeland, R.A.; Tummino, P.J.; Luo, L. Epigallocatechin gallate (EGCG), a major component of green tea, is a dual phosphoinositide-3-kinase/mTOR inhibitor. Biochem. Biophys. Res. Commun. 2011, 406, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, Y.; Jia, C.; Wang, Y.; Lai, P.; Zhou, X.; Wang, Y.; Song, Q.; Lin, J.; Ren, Z.; et al. mTORC1/2 targeted by n-3 polyunsaturated fatty acids in the prevention of mammary tumorigenesis and tumor progression. Oncogene 2014, 33, 4548–4557. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, X.; Guo, Y.; Chan, L.; Guan, X. α-Lipoic acid increases energy expenditure by enhancing adenosine monophosphate–activated protein kinase–peroxisome proliferator-activated receptor-γ coactivator-1α signaling in the skeletal muscle of aged mice. Metabolism 2010, 59, 967–976. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef]
- Reznick, R.M.; Zong, H.; Li, J.; Morino, K.; Moore, I.K.; Yu, H.J.; Liu, Z.-X.; Dong, J.; Mustard, K.J.; Hawley, S.A.; et al. Aging-Associated Reductions in AMP-Activated Protein Kinase Activity and Mitochondrial Biogenesis. Cell Metab. 2007, 5, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Ljubicic, V.; Hood, D.A. Diminished contraction-induced intracellular signaling towards mitochondrial biogenesis in aged skeletal muscle. Aging Cell 2009, 8, 394–404. [Google Scholar] [CrossRef]
- Poels, J.; Spasić, M.R.; Callaerts, P.; Norga, K.K. Expanding roles for AMP-activated protein kinase in neuronal survival and autophagy. Bioessays 2009, 31, 944–952. [Google Scholar] [CrossRef]
- Yang, T.Y.T.; Wang, L.W.L.; Zhu, M.Z.M.; Yan, L.Y.L. Properties and molecular mechanisms of resveratrol: A review. Die Pharm. -Int. J. Pharm. Sci. 2015, 70, 501–506. [Google Scholar] [CrossRef]
- Haigis, M.C.; Sinclair, D.A. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 253–295. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, K.; Chaudhary, G.; Gupta, Y.K. Protective effect of resveratrol against oxidative stress in middle cerebral artery occlusion model of stroke in rats. Life Sci. 2002, 71, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xu, J.; Rottinghaus, G.E.; Simonyi, A.; Lubahn, D.; Sun, G.Y.; Sun, A.Y. Resveratrol protects against global cerebral ischemic injury in gerbils. Brain Res. 2002, 958, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Amri, A.; Chaumeil, J.C.; Sfar, S.; Charrueau, C. Administration of resveratrol: What formulation solutions to bioavailability limitations? J. Control. Release 2012, 158, 182–193. [Google Scholar] [CrossRef]
- Sarubbo, F.; Moranta, D.; Asensio, V.J.; Miralles, A.; Esteban, S. Effects of Resveratrol and Other Polyphenols on the Most Common Brain Age-Related Diseases. Curr. Med. Chem. 2017, 24, 4245–4266. [Google Scholar] [CrossRef] [PubMed]
- Cosín-Tomàs, M.; Senserrich, J.; Arumí-Planas, M.; Alquézar, C.; Pallàs, M.; Martín-Requero, Á.; Suñol, C.; Kaliman, P.; Sanfeliu, C. Role of Resveratrol and Selenium on Oxidative Stress and Expression of Antioxidant and Anti-Aging Genes in Immortalized Lymphocytes from Alzheimer’s Disease Patients. Nutrients 2019, 11, 1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.-C.; Guarente, L. SIRT1 Mediates Central Circadian Control in the SCN by a Mechanism that Decays with Aging. Cell 2013, 153, 1448–1460. [Google Scholar] [CrossRef] [Green Version]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian Control of the NAD+ Salvage Pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef]
- Braidy, N.; Berg, J.; Clement, J.; Khorshidi, F.; Poljak, A.; Jayasena, T.; Grant, R.; Sachdev, P. Role of nicotinamide sdenine dinucleotide and related precursors as therapeutic targets for age-related degenerative diseases: Rationale, biochemistry, pharmacokinetics, and outcomes. Antioxid. Redox Signal. 2019, 30, 251–294. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, K.D.; Connor, C.M.; Campan, M.; Long, T.I.; Weisenberger, D.J.; Biniszkiewicz, D.; Jaenisch, R.; Laird, P.W.; Akbarian, S. DNA Methylation in the Human Cerebral Cortex Is Dynamically Regulated throughout the Life Span and Involves Differentiated Neurons. PLoS ONE 2007, 2, e895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, R.; Thomas, P.; Zalewski, P.; Fenech, M. Zinc supplementation influences genomic stability biomarkers, antioxidant activity, and zinc transporter genes in an elderly Australian population with low zinc status. Mol. Nutr. Food Res. 2015, 59, 1200–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aaseth, J.; Alexander, J.; Bjørklund, G.; Hestad, K.; Dusek, P.; Roos, P.M.; Alehagen, U. Treatment strategies in Alzheimer’s disease: A review with focus on selenium supplementation. Biometals 2016, 29, 827–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macaron, T.; Giudici, K.V.; Bowman, G.L.; Sinclair, A.; Stephan, E.; Vellas, B.; Barreto, P.D.S. Associations of Omega-3 fatty acids with brain morphology and volume in cognitively healthy older adults: A narrative review. Ageing Res. Rev. 2021, 67, 101300. [Google Scholar] [CrossRef]
- Lin, P.-Y.; Chiu, C.-C.; Huang, S.-Y.; Su, K.-P. A Meta-Analytic Review of Polyunsaturated Fatty Acid Compositions in Dementia. J. Clin. Psychiatry 2012, 73, 1245–1254. [Google Scholar] [CrossRef]
- Bo, Y.; Zhang, X.; Wang, Y.; You, J.; Cui, H.; Zhu, Y.; Pang, W.; Liu, W.; Jiang, Y.; Lu, Q. The n-3 Polyunsaturated Fatty Acids Supplementation Improved the Cognitive Function in the Chinese Elderly with Mild Cognitive Impairment: A Double-Blind Randomized Controlled Trial. Nutrients 2017, 9, 54. [Google Scholar] [CrossRef] [Green Version]
- Avallone, R.; Vitale, G.; Bertolotti, M. Omega-3 Fatty Acids and Neurodegenerative Diseases: New Evidence in Clinical Trials. Int. J. Mol. Sci. 2019, 20, 4256. [Google Scholar] [CrossRef] [Green Version]
- White III, C.W.; Fan, X.; Maynard, J.C.; Wheatley, E.G.; Bieri, G.; Couthouis, J.; Burlingame, A.L.; Villeda, S.A. Age-related loss of neural stem cell O-GlcNAc promotes a glial fate switch through STAT3 activation. Proc. Natl. Acad. Sci. USA 2020, 117, 22214–22224. [Google Scholar] [CrossRef]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef]
- Moradi, H.R.; Hajali, V.; Khaksar, Z.; Vafaee, F.; Forouzanfar, F.; Negah, S.S. The next step of neurogenesis in the context of Alzheimer’s disease. Mol. Biol. Rep. 2021, 48, 5647–5660. [Google Scholar] [CrossRef] [PubMed]
- Akan, I.; Halim, A.; Vakhrushev, S.; Clausen, H.; Hanover, J. Drosophila O-GlcNAcase Mutants Reveal an Expanded Glycoproteome and Novel Growth and Longevity Phenotypes. Cells 2021, 10, 1026. [Google Scholar] [CrossRef] [PubMed]
- Colcombe, S.J.; Kramer, A.F. Fitness effects on the cognitive function of older adults: A meta-analytic study. Psychol. Sci. 2003, 14, 125–130. [Google Scholar] [CrossRef]
- Colcombe, S.J.; Erickson, K.I.; Raz, N.; Webb, A.G.; Cohen, N.J.; McAuley, E.; Kramer, A.F. Aerobic Fitness Reduces Brain Tissue Loss in Aging Humans. J. Gerontol. Ser. A 2003, 58, M176–M180. [Google Scholar] [CrossRef] [Green Version]
- Piepmeier, A.T.; Etnier, J.L. Brain-derived neurotrophic factor (BDNF) as a potential mechanism of the effects of acute exercise on cognitive performance. J. Sport Health Sci. 2015, 4, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Khan, R.; Cortes, C.J. Forgot to Exercise? Exercise Derived Circulating Myokines in Alzheimer’s Disease: A Perspective. Front. Neurol. 2021, 12, 649452. [Google Scholar] [CrossRef]
- Willcox, B.J.; Willcox, D.C. Caloric restriction, caloric restriction mimetics, and healthy aging in Okinawa: Controversies and clinical implications. Curr. Opin. Clin. Nutr. Metab. Care. 2014, 17, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Weindruch, R.; Walford, R.L. The Retardation of Aging and Disease by Dietary Restriction; Charles, C., Ed.; Thomas Pub. Ltd.: Springfield, IL, USA, 1988. [Google Scholar]
- Anderson, R.M.; Le Couteur, D.G.; De Cabo, R. Caloric Restriction Research: New Perspectives on the Biology of Aging. J. Gerontol. Ser. A 2017, 73, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwangbo, D.-S.; Lee, H.-Y.; Abozaid, L.S.; Min, K.-J. Mechanisms of Lifespan Regulation by Calorie Restriction and Intermittent Fasting in Model Organisms. Nutrients 2020, 12, 1194. [Google Scholar] [CrossRef]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef]
- Neumann, B.; Baror, R.; Zhao, C.; Segel, M.; Dietmann, S.; Rawji, K.S.; Foerster, S.; McClain, C.R.; Chalut, K.; van Wijngaarden, P.; et al. Metformin restores CNS remyelination capacity by rejuvenating aged stem cells. Cell Stem. Cell 2019, 25, 473–485.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, P.; Saarma, M. Novel CDNF/MANF family of neurotrophic factors. Dev. Neurobiol. 2010, 70, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, P.; Peränen, J.; Andressoo, J.-O.; Kalkkinen, N.; Kokaia, Z.; Lindvall, O.; Timmusk, T.; Saarma, M. MANF is widely expressed in mammalian tissues and differently regulated after ischemic and epileptic insults in rodent brain. Mol. Cell. Neurosci. 2008, 39, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yang, H.; Chang, R.; Yin, P.; Yang, Y.; Yang, W.; Huang, S.; Gaertig, M.A.; Li, S.; Li, X.-J. MANF regulates hypothalamic control of food intake and body weight. Nat. Commun. 2017, 8, 579. [Google Scholar] [CrossRef] [Green Version]
- Sousa-Victor, P.; Neves, J.; Cedron-Craft, W.; Ventura, P.B.; Liao, C.-Y.; Riley, R.R.; Soifer, I.; van Bruggen, N.; Kolumam, G.A.; Villeda, S.A.; et al. MANF regulates metabolic and immune homeostasis in ageing and protects against liver damage. Nat. Metab. 2019, 1, 276–290. [Google Scholar] [CrossRef]
- Poehlman, E.T.; Turturro, A.; Bodkin, N.; Cefalu, W.; Heymsfield, S.; Holloszy, J.; Kemnitz, J. Caloric Restriction Mimetics: Physical Activity and Body Composition Changes. J. Gerontol. Ser. A 2001, 56, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Carey, A.L.; Kingwell, B.A. Novel pharmacological approaches to combat obesity and insulin resistance: Targeting skeletal muscle with ‘exercise mimetics’. Diabetologia 2009, 52, 2015–2026. [Google Scholar] [CrossRef]
- Goldman, D.P.; Cutler, D.; Rowe, J.W.; Michaud, P.-C.; Sullivan, J.; Peneva, D.; Olshansky, S.J. Substantial Health And Economic Returns From Delayed Aging May Warrant A New Focus For Medical Research. Health Aff. 2013, 32, 1698–1705. [Google Scholar] [CrossRef] [Green Version]
- Goldman, D. The economic returns to delayed aging: Promise and pitfalls. Innov. Aging 2017, 1, 1082. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xu, S.; Yu, M.; Wang, K.; Tao, Y.; Zhou, Y.; Shi, J.; Zhou, M.; Wu, B.; Yang, Z.; et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J. Allergy Clin. Immunol. 2020, 146, 110–118. [Google Scholar] [CrossRef]
- Bischof, E.; Siow, R.C.; Zhavoronkov, A.; Kaeberlein, M. The potential of rapalogs to enhance resilience against SARS-CoV-2 infection and reduce the severity of COVID-19. Lancet 2021, 2, E105–E111. [Google Scholar] [CrossRef] [PubMed]
- Galkin, F.; Parish, A.; Bischof, E.; Zhang, J.; Mamoshina, P.; Zhavoronkov, A. Increased Pace of Aging in COVID-Related Mortality. Life 2021, 11, 730. [Google Scholar] [CrossRef] [PubMed]
- Olshansky, S.J.; Goldman, D.P.; Zheng, Y.; Rowe, J.W. Aging in America in the Twenty-first Century: Demographic Forecasts from the MacArthur Foundation Research Network on an Aging Society. Milbank Q. 2009, 87, 842–862. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.K. New Methuselahs: The Ethics of Life Extension; MIT Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Pyrkov, T.V.; Avchaciov, K.; Tarkhov, A.E.; Menshikov, L.I.; Gudkov, A.V.; Fedichev, P.O. Longitudinal analysis of blood markers reveals progressive loss of resilience and predicts human lifespan limit. Nat. Commun. 2021, 12, 2765. [Google Scholar] [CrossRef]
- Armstrong, N.J.; Mather, K.A.; Thalamuthu, A.; Wright, M.J.; Trollor, J.N.; Ames, D.; Brodaty, H.; Schofield, P.R.; Sachdev, P.S.; Kwok, J.B. Aging, exceptional longevity and comparisons of the Hannum and Horvath epigenetic clocks. Epigenomics 2017, 9, 689–700. [Google Scholar] [CrossRef]
- Demidenko, O.; Barardo, D.; Budovskii, V.; Finnemore, R.; Palmer, F.R.; Kennedy, B.K.; Budovskaya, Y.V. Rejuvant®, a potential life-extending compound formulation with alpha-ketoglutarate and vitamins, conferred an average 8 year reduction in biological aging, after an average of 7 months of use, in the TruAge DNA methylation test. Aging 2021, 13, 24485–24499. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, R.E.; Heiens, R.A. Subjective age: A test of five hypotheses. Gerontologist 1992, 32, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Westerhof, G. Forever young? A comparison of age identities in the United States and Germany. Res. Aging 2003, 25, 366–383. [Google Scholar] [CrossRef] [Green Version]
- Montepare, J.M.; Lachman, M.E. “You’re only as old as you feel”: Self-perceptions of age, fears of aging, and life satisfaction from adolescence to old age. Psychol. Aging 1989, 4, 73–78. [Google Scholar] [CrossRef]
- Weiss, D.; Lang, F.R. “They” are old but “I” feel younger: Age-group dissociation as a self-protective strategy in old age. Psychol. Aging 2012, 27, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, M.J.; Sachdev, P. Brain reserve and dementia: A systematic review. Psychol. Med. 2006, 36, 441–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, H.; Anstey, K.J.; Leach, L.S.; Mackinnon, A.J. Intelligence, Education, and the Brain Reserve Hypothesis. In The Handbook of Aging and Cognition, 3rd ed.; Craik, F.I.M., Salthouse, T.A., Eds.; Psychology Press: New York, NY, USA, 2008; pp. 133–188. [Google Scholar]
- Garibotto, V.; Borroni, B.; Sorbi, S.; Cappa, S.F.; Padovani, A.; Perani, D. Education and occupation provide reserve in both ApoE ε4 carrier and noncarrier patients with probable Alzheimer’s disease. Neurol. Sci. 2012, 33, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.W.; Stepanovic, M.R.; Andreano, J.; Barrett, L.F.; Touroutoglou, A.; Dickerson, B.C. Youthful Brains in Older Adults: Preserved Neuroanatomy in the Default Mode and Salience Networks Contributes to Youthful Memory in Superaging. J. Neurosci. 2016, 36, 9659–9668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balduino, E.; de Melo, B.A.R.; Silva, L.D.S.M.D.; Martinelli, J.E.; Cecato, J.F. The “SuperAgers” construct in clinical practice: Neuropsychological assessment of illiterate and educated elderly. Int. Psychogeriatr. 2020, 32, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.H.; Sridhar, J.; Ohm, D.; Rademaker, A.; Mesulam, M.-M.; Weintraub, S.; Rogalski, E. Rates of Cortical Atrophy in Adults 80 Years and Older With Superior vs Average Episodic Memory. JAMA 2017, 317, 1373–1375. [Google Scholar] [CrossRef] [Green Version]
- Rogalski, E.J.; Gefen, T.; Shi, J.; Samimi, M.; Bigio, E.; Weintraub, S.; Geula, C.; Mesulam, M.-M. Youthful Memory Capacity in Old Brains: Anatomic and Genetic Clues from the Northwestern SuperAging Project. J. Cogn. Neurosci. 2013, 25, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Brody, A.L.; Mandelkern, M.A.; Jarvik, M.E.; Lee, G.S.; Smith, E.C.; Huang, J.C.; London, E.D. Differences between smokers and nonsmokers in regional gray matter volumes and densities. Biol. Psychiat. 2004, 55, 77–84. [Google Scholar] [CrossRef]
- Almeida, O.P.; Garrido, G.J.; Lautenschlager, N.T.; Hulse, G.K.; Jamrozik, K.; Flicker, L. Smoking Is Associated With Reduced Cortical Regional Gray Matter Density in Brain Regions Associated With Incipient Alzheimer Disease. Am. J. Geriatr. Psychiatry 2008, 16, 92–98. [Google Scholar] [CrossRef]
- Pfefferbaum, A.; Lim, K.O.; Zipursky, R.B.; Mathalon, D.H.; Rosenbloom, M.J.; Lane, B.; Ha, C.N.; Sullivan, E.V. Brain Gray and White Matter Volume Loss Accelerates with Aging in Chronic Alcoholics: A Quantitative MRI Study. Alcohol. Clin. Exp. Res. 1992, 16, 1078–1089. [Google Scholar] [CrossRef]
- Ning, K.; Zhao, L.; Matloff, W.; Sun, F.; Toga, A.W. Association of relative brain age with tobacco smoking, alcohol consumption, and genetic variants. Sci. Rep. 2020, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Scarmeas, N.; Short, E.E.; Luchsinger, J.A.; DeCarli, C.; Stern, Y.; Manly, J.J.; Schupf, N.; Mayeux, R.; Brickman, A.M. Alcohol intake and brain structure in a multiethnic elderly cohort. Clin. Nutr. 2014, 33, 662–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reale, M.; Costantini, E.; Jagarlapoodi, S.; Khan, H.; Belwal, T.; Cichelli, A. Relationship of Wine Consumption with Alzheimer’s Disease. Nutrients 2020, 12, 206. [Google Scholar] [CrossRef] [Green Version]
- Dobrohotova, T.A.; Bragina, N.N. The Left-Handers; Kniga: Moscow, Russia, 1994. [Google Scholar]
- Marks, J.S.; Williamson, D.F. Left-handedness and life expectancy. N. Engl. J. Med. 1991, 325, 1041–1043. [Google Scholar]
- Harris, L.J. Do left-handers die sooner than right-handers? Commentary on Coren and Halpern’s (1991) “Left-handedness: A marker for decreased survival fitness”. Psychol. Bull. 1993, 114, 203–234. [Google Scholar] [CrossRef] [PubMed]
- de Leon, M.J.; la Regina, M.E.; Ferris, S.H.; Gentes, C.I.; Miller, J.D. Reduced incidence of left-handedness in clinically diagnosed dementia of the Alzheimer type. Neurobiol. Aging 1986, 7, 161–164. [Google Scholar] [CrossRef]
- Doody, R.S.; Vacca, J.L.; Massman, P.J.; Liao, T.-Y. The Influence of Handedness on the Clinical Presentation and Neuropsychology of Alzheimer Disease. Arch. Neurol. 1999, 56, 1133–1137. [Google Scholar] [CrossRef] [Green Version]
- Ruigrok, A.N.; Salimi-Khorshidi, G.; Lai, M.-C.; Baron-Cohen, S.; Lombardo, M.V.; Tait, R.J.; Suckling, J. A meta-analysis of sex differences in human brain structure. Neurosci. Biobehav. Rev. 2014, 39, 34–50. [Google Scholar] [CrossRef] [Green Version]
- Kiesow, H.; Dunbar, R.I.M.; Kable, J.W.; Kalenscher, T.; Vogeley, K.; Schilbach, L.; Marquand, A.F.; Wiecki, T.V.; Bzdok, D. 10,000 social brains: Sex differentiation in human brain anatomy. Sci. Adv. 2020, 6, eaaz1170. [Google Scholar] [CrossRef] [Green Version]
- Jiang, R.; Calhoun, V.D.; Fan, L.; Zuo, N.; Jung, R.; Qi, S.; Lin, D.; Li, J.; Zhuo, C.; Song, M.; et al. Gender Differences in Connectome-based Predictions of Individualized Intelligence Quotient and Sub-domain Scores. Cereb. Cortex 2020, 30, 888–900. [Google Scholar] [CrossRef] [Green Version]
- Coffey, C.; Lucke, J.; Saxton, J.; Ratcliff, G.; Unitas, L.; Billig, B.; Bryan, R.N. Sex differences in brain aging: A quantitative magnetic resonance imaging study. Arch Neurol. 1998, 55, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Kobayashi, S.; Yamaguchi, S.; Iijima, K.-I.; Okada, K.; Yamashita, K. Gender Effects on Age-Related Changes in Brain Structure. Am. J. Neuroradiol. 2000, 21, 112–118. [Google Scholar] [PubMed]
- Ngun, T.C.; Ghahramani, N.; Sánchez, F.J.; Bocklandt, S.; Vilain, E. The genetics of sex differences in brain and behavior. Front. Neuroendocr. 2011, 32, 227–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trabzuni, D.; Ramasamy, A.; Imran, S.; Walker, R.; Smith, C.; Weale, M.E.; Hardy, J.; Ryten, M. North American Brain Expression Consortium Widespread sex differences in gene expression and splicing in the adult human brain. Nat. Commun. 2013, 4, 2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, E.; Lamport, D.; Brennan, L.; Burnet, P.; Calabrese, V.; Cunnane, S.C.; de Wilde, M.C.; Dye, L.; Farrimond, J.A.; Lombardo, N.E.; et al. Nutrition and the ageing brain: Moving towards clinical applications. Ageing Res. Rev. 2020, 62, 101079. [Google Scholar] [CrossRef] [PubMed]







| Characteristics | BBA > CA | BBA < CA | p-Value | Test Type |
|---|---|---|---|---|
| Sample size (N) | 28 | 61 | Not applicable | Not applicable |
| Sex (% of females) | 57.1 | 65.6 | Not significant | Chi-square |
| Chronological age—CA (mean/st.d) | 42.2 (8.9) | 52.7 (10.7) | 0.00001 | Mann–Whitney U test |
| Brain biological age—BBA (mean/st.d) | 51.4 (7.3) | 37.3 (9.9) | 0.00001 | Mann–Whitney U test |
| Brain resources—BR (mean %/st.d) | −10.5 (9.2) | +18.6 (11.0) | 0.00001 | Mann–Whitney U test |
| Healthy lifestyle habits (% of those who have) | 14.3 | 14.8 | Not significant | Chi-square |
| Current health symptoms (% of those who have) | 42.9 | 60.7 | 0.010846 | Chi-square |
| Past health problems (% of those who had) | 53.6 | 62.3 | Not significant | Chi-square |
| Relatives with mind/brain disorders (% of those who have) | 17.9 | 23.1 | Not significant | Chi-square |
| Anxiety–Beck (mean/st.d) | 7.0 (6.5) | 8.5 (6.8) | Not significant | Mann–Whitney U test |
| Anxiety–Ham (mean/st.d) | 7.3 (5.8) | 9.2 (6.0) | Not significant | Mann–Whitney U test |
| Depression–Beck (mean/st.d) | 5.6 (5.3) | 6.6 (5.9) | Not significant | Mann–Whitney U test |
| Big-5—neuroticism (mean/st.d) | 2.7 (0.8) | 2.9 (0.7) | Not significant | Mann–Whitney U test |
| Handedness (% of right-handed) | 92.8 | 83.6 | 0.046061 | Chi-square |
| Marital status (% of married) | 66 | 82 | 0.0099 | Chi-square |
| Marital status (% of divorced) | 19.7 | 11.5 | Not significant | Chi-square |
| Marital status (% of single) | 14.6 | 6.5 | 0.037897 | Chi-square |
| Education (% of those who have a PhD) | 3.7 | 16.4 | 0.004678 | Chi-square |
| Education (% of those who graduated from university or institute) | 67.8 | 75.4 | Not significant | Chi-square |
| Education (% of those who completed high school (≥11–12 years)) | 28.5 | 8.2 | 0.000131 | Chi-square |
| Job (% of directors or CEOs) | 28.6 | 21.3 | Not significant | Chi-square |
| Job (% of senior managers) | 17.9 | 23.1 | Not significant | Chi-square |
| Job (% of junior managers) | 46.4 | 54 | Not significant | Chi-square |
| Job (% of students or trainees) | 7.1 | 1.6 | Not significant | Chi-square |
| Number of interests or hobbies (mean/st.d) | 3.0 (1.3) | 4.5 (1.6) | 0.00026 | Mann–Whitney U test |
| Smoking (% of those who smoke) | 7.1 | 1.2 | 0.030383 | Chi-square |
| Alcohol consumption (1–2 drinks per week; %) | 46.4 | 47.5 | Not significant | Chi-square |
| Alcohol consumption (3–4 drinks per week; %) | 35.7 | 34.4 | Not significant | Chi-square |
| Alcohol consumption (5–7 drinks per week; %) | 14.3 | 6.6 | Not significant | Chi-square |
| Alcohol consumption (8–10 drinks per week; %) | 3.6 | 11.5 | 0.037056 | Chi-square |
| Groups | Pre-Intervention | Postintervention |
|---|---|---|
| Experimental/nutraceuticals | 33.3 | 4.7 |
| Control/lifestyle | 40.2 | 32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fingelkurts, A.A.; Fingelkurts, A.A. Turning Back the Clock: A Retrospective Single-Blind Study on Brain Age Change in Response to Nutraceuticals Supplementation vs. Lifestyle Modifications. Brain Sci. 2023, 13, 520. https://doi.org/10.3390/brainsci13030520
Fingelkurts AA, Fingelkurts AA. Turning Back the Clock: A Retrospective Single-Blind Study on Brain Age Change in Response to Nutraceuticals Supplementation vs. Lifestyle Modifications. Brain Sciences. 2023; 13(3):520. https://doi.org/10.3390/brainsci13030520
Chicago/Turabian StyleFingelkurts, Andrew A., and Alexander A. Fingelkurts. 2023. "Turning Back the Clock: A Retrospective Single-Blind Study on Brain Age Change in Response to Nutraceuticals Supplementation vs. Lifestyle Modifications" Brain Sciences 13, no. 3: 520. https://doi.org/10.3390/brainsci13030520