
The Role of Autophagy and lncRNAs in the Maintenance of Cancer Stem Cells
 ,
,  ,
,  , , and
, , and 
Abstract
:Simple Summary
Abstract

1. Introduction
2. Methods for Detecting and Understanding the Characteristics of CSCs
3. Identification of Cancer Stem Cells in an Array of Cancers
3.1. Acute Myeloid Leukaemia (AML)
3.2. Oesophageal Cancer
3.3. Colorectal Cancer
3.4. Gastric Cancer
3.5. Pancreatic Cancer
3.6. Hepatocellular Carcinoma
3.7. Lung Cancer
3.8. Glioblastoma Multiforme
3.9. Osteosarcoma
3.10. Breast Cancer
4. Autophagy and CSCs
4.1. Autophagy: A Cellular Pro-Survival Process
4.2. CSCs Rely on Autophagy for Stemness, Invasion, Migration, and Chemo-Resistance
4.3. Autophagy and the Hypoxic Microenvironment
4.4. CSCs Are Dependent on Mitophagy for Their Metabolic Reprogramming
5. Long Noncoding RNAs and Cancer Stem Cells
5.1. Long Noncoding RNAs in Health and Disease: A Central Role in Cancer
5.2. LncRNAs in CSC Maintenance and Migration
5.2.1. Epigenetic Regulators
5.2.2. miRNA Synthesis and Function
5.2.3. Transcriptional Regulators
5.2.4. LncRNA-Mediated Regulation of CSCs Modulates Drug Resistance
5.3. LncRNAs as Cancer and CSC-Specific Therapeutic Targets and Biomarkers
6. The Interrogation of Gene–Disease Networks for CSC-Associated Genes
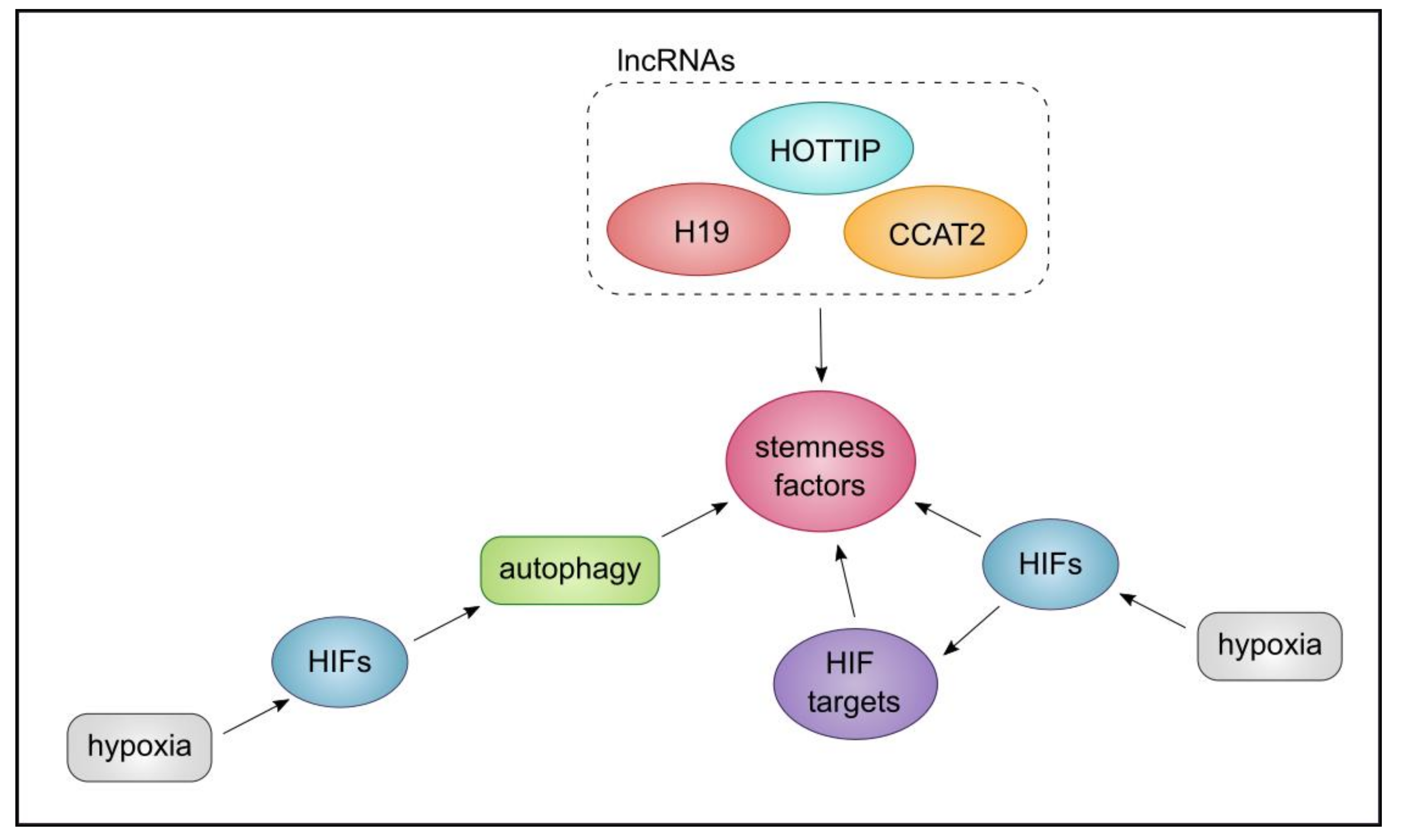
7. The Triangle of CSCs, Autophagy, and lncRNA
8. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CSCs | Cancer stem cells |
| TICs | Tumour initiating cells |
| LncRNA | Long noncoding RNA |
| TME | Tumour microenvironment |
| NOD/SCID | Nonobese diabetic/severe combined immunodeficient mice model |
| ROS | Reactive oxygen species |
| AML | Acute myeloid leukaemia |
| ABC | ATP-binding cassette transporter |
| SP | Side population |
| T-ALL | T-cell acute lymphoblastic leukaemia |
| MTS | Multicellular tumour spheroid |
| RA (ATRA) | All-trans retinoic acid |
| ALDH1 | Aldehyde dehydrogenase 1 |
| MMP | Matrix metalloproteinase |
| PI3K/AKT pathway | Phosphoinositide 3-kinase/protein kinase |
| EGFR | Epidermal growth factor receptor |
| ESA | Epithelial-specific antigen |
| CD | Cluster of differentiation |
| EPCAM | Epithelial cell adhesion molecule |
| BCSCs | Breast cancer stem cells |
| UPS | Ubiquitin proteasome system |
| ATGs | Autophagy-related proteins |
| EMT | Epithelial-to-mesenchymal transition |
| 3-MA | 3-methyladenine |
| HIF-1α | Hypoxia-inducible factor 1alpha |
| DRP1 | Dynamin-related protein 1 |
| KMT2C | Lysine (K)-specific methyltransferase 2C |
| LTR | Long terminal repeat |
| 5-FU | 5-fluorouracil |
| STAT3 | Signal transducer and activator of transcription 3 |
| CML | Chronic myelogenous leukaemia |
| TFs | Transcription factors |
| LSCs | Leukaemic stem cells |
| TKI | Tyrosine Kinase inhibitor |
| DNMT3A | DNA methyltransferase 3A |
| HDAC1 | Histone deacetylase 1 |
| ALL | Acute lymphoblastic leukaemia |
| IL-6 | Interleukin 6 |
References
- Yap, T.A.; Futreal, P.A.; Pusztai, L.; Swanton, C.; Gerlinger, M. Intratumor heterogeneity: Seeing the wood for the trees. Sci. Transl. Med. 2012, 4, 127ps10. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M.; Maley, C.C. Clonal evolution in Cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Allan, A.L.; Vantyghem, S.A.; Tuck, A.B.; Chambers, A.F. Tumor dormancy and cancer stem cells: Implications for the biology and treatment of breast cancer metastasis. Breast Dis. 2006, 26, 87–98. [Google Scholar] [CrossRef]
- Cho, R.W.; Clarke, M.F. Recent advances in cancer stem cells. Curr. Opin. Genet. Dev. 2008, 18, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Berry, J.E.; Eber, M.R.; Jung, Y.; Yumoto, K.; Cackowski, F.C.; Yoon, H.J.; Parsana, P.; Mehra, R.; Wang, J.; et al. The marrow niche controls the cancer stem cell phenotype of disseminated prostate. Cancer Oncotarget 2016, 7, 41217–41232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Huntly, B.J.; Gilliland, D.G. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat. Rev. Cancer 2005, 4, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal Cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. PNAS 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells–perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Bruttel, V.S.; Wischhusen, J. Cancer stem cell immunology: Key to understanding tumorigenesis and tumor immune escape? Front. Immunol. 2014, 5, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallette, F.M.; Olivier, C.; Lézot, F.; Oliver, L.; Cochonneau, D.; Lalier, L.; Cartron, P.F.; Heymann, D. Dormant, quiescent, tolerant and persister cells: Four synonyms for the same target in Cancer. Biochem. Pharmacol. 2019, 162, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Rycaj, K.; Tang, D.G. Cell-of-Origin of Cancer versus Cancer Stem Cells: Assays and Interpretations. Cancer Res. 2015, 75, 4003–4011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell 2012, 22, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Eades, G.; Zhang, Y.S.; Li, Q.L.; Xia, J.X.; Yao, Y.; Zhou, Q. Long non-coding RNAs in stem cells and Cancer. World J. Clin. Oncol. 2014, 5, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; He, L.; Du, Y.; Zhu, P.; Huang, G.; Luo, J.; Yan, X.; Ye, B.; Li, C.; Xia, P.; et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell. 2015, 16, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Oropeza, R.; Melendez-Zajgla, J.; Maldonado, V.; Vazquez-Santillan, K. The emerging role of lncRNAs in the regulation of cancer stem cells. Cell Oncol. 2018, 41, 585–603. [Google Scholar] [CrossRef]
- Lecerf, C.; Le Bourhis, X.; Adriaenssens, E. The long non-coding RNA H19: An active player with multiple facets to sustain the hallmarks of Cancer. Cell Mol. Life Sci. 2019, 76, 4673–4687. [Google Scholar] [CrossRef] [PubMed]
- Zaarour, R.F.; Azakir, B.; Hajam, E.Y.; Nawafleh, H.; Zeinelabdin, N.A.; Engelsen, A.S.T.; Thiery, J.; Jamora, C.; Chouaib, S. Role of Hypoxia-Mediated Autophagy in Tumor Cell Death and Survival. Cancers 2021, 13, 503. [Google Scholar] [CrossRef] [PubMed]
- Hanekamp, D.; Cloos, J.; Schuurhuis, G.J. Leukemic stem cells: Identification and clinical application. Int. J. Hematol. 2017, 105, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Dick, J.E. Acute myeloid leukemia stem cells. Ann. N. Y. Acad. Sci. 2005, 1044, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Terpstra, W.; Prins, A.; Ploemacher, R.E.; Wognum, B.W.; Wagemaker, G.; Löwenberg, B.; Wielenga, J.J. Long-term leukemia-initiating capacity of a CD34− subpopulation of acute myeloid leukemia. Blood 1996, 87, 2187–2194. [Google Scholar] [CrossRef] [Green Version]
- Piao, L.; Yang, Z.; Jin, J.; Ni, W.; Qi, W.; Xuan, Y. B7H4 is associated with stemness and cancer progression in esophageal squamous cell carcinoma. Hum. Pathol. 2018, 80, 152–162. [Google Scholar] [CrossRef]
- Yang, Z.; Ni, W.; Cui, C.; Qi, W.; Piao, L.; Xuan, Y. Identification of LETM1 as a marker of cancer stem-like cells and predictor of poor prognosis in esophageal squamous cell carcinoma. Hum. Pathol. 2018, 81, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Hirohashi, Y.; Murai, A.; Nishidate, T.; Okita, K.; Wang, L.; Ikehara, Y.; Satoyoshi, T.; Usui, A.; Kubo, T.; et al. ST6GALNAC1 plays important roles in enhancing cancer stem phenotypes of colorectal cancer via the Akt pathway. Oncotarget 2017, 8, 112550–112564. [Google Scholar] [CrossRef] [Green Version]
- Xue, Z.; Yan, H.; Li, J.; Liang, S.; Cai, X.; Chen, X.; Wu, Q.; Gao, L.; Wu, K.; Nie, Y.; et al. Identification of cancer stem cells in vincristine preconditioned SGC7901 gastric cancer cell line. J. Cell Biochem. 2012, 113, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Xu, J.; Li, L.; Ye, W.; Xu, G.; Chen, B.; Zeng, J.; Li, J.; Huang, Z. Effect of gastric cancer stem cell on gastric cancer invasion, migration and angiogenesis. Int. J. Med. Sci. 2020, 17, 2040–2051. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic Cancer. Cell Stem Cell. 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, T.; Wang, X.W. Cancer stem cells in the development of liver Cancer. J. Clin. Invest. 2013, 123, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Maiorana, A.; Sighinolfi, P.; Stefani, A.; Morandi, U.; Dominici, M.; Aramini, B. Isolation and Identification of Cancer Stem-Like Cells in Adenocarcinoma and Squamous Cell Carcinoma of the Lung: A Pilot Study. Bd. 9. Front. Oncol. 2019, s, 1394. [Google Scholar] [CrossRef] [Green Version]
- Herreros-Pomares, A.; De-Maya-Girones, J.D.; Calabuig-Fariñas, S.; Lucas, R.; Martínez, A.; Pardo-Sánchez, J.M.; Alonso, S.; Blasco, A.; Guijarro, R.; Martorell, M.; et al. Lung tumorspheres reveal cancer stem cell-like properties and a score with prognostic impact in resected non-small-cell lung. Cancer Cell Death Dis. 2019, 10, 660. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sakariassen, P.Ø.; Tsinkalovsky, O.; Immervoll, H.; Bøe, S.O.; Svendsen, A.; Prestegarden, L.; Røsland, G.; Thorsen, F.; Stuhr, L.; et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int. J. Cancer 2008, 122, 761–768. [Google Scholar] [CrossRef]
- Bradshaw, A.; Wickremesekera, A.; Brasch, H.D.; Chibnall, A.M.; Davis, P.F.; Tan, S.T.; Itinteang, T. Cancer Stem Cells in Glioblastoma Multiforme. Front. Surg. 2016, 348, 48. [Google Scholar] [CrossRef] [Green Version]
- Basu-Roy, U.; Seo, E.; Ramanathapuram, L.; Rapp, T.B.; Perry, J.A.; Orkin, S.H.; Mansukhani, A.; Basilico, C. Sox2 maintains self renewal of tumor-initiating cells in osteosarcomas. Oncogene 2012, 31, 2270–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, M.P.; da Conceição Braga, L.; Silva, L.M. STAT3 as a promising chemoresistance biomarker associated with the CD44+/high/CD24-/low/ALDH+ BCSCs-like subset of the triple-negative breast cancer (TNBC) cell line. Exp. Cell Res. 2018, 363, 283–290. [Google Scholar] [CrossRef]
- Golebiewska, A.; Brons, N.H.; Bjerkvig, R.; Niclou, S.P. Critical appraisal of the side population assay in stem cell and cancer stem cell research. Cell Stem Cell. 2011, 8, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Murase, M.; Kano, M.; Tsukahara, T.; Takahashi, A.; Torigoe, T.; Kawaguchi, S.; Kimura, S.; Wada, T.; Uchihashi, Y.; Kondo, T.; et al. Side population cells have the characteristics of cancer stem-like cells/cancer-initiating cells in bone sarcomas. Br. J. Cancer 2009, 101, 1425–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and Cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Chai, S.; Wang, P.; Zhang, C.; Yang, Y.; Yang, Y.; Wang, K. Aldehyde dehydrogenases and cancer stem cells. Cancer Lett. 2015, 369, 50–57. [Google Scholar] [CrossRef]
- Somervaille, T.C.; Cleary, M.L. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006, 10, 257–268. [Google Scholar] [CrossRef]
- Liu, L.; Cheung, T.H.; Charville, G.W.; Hurgo, B.M.; Leavitt, T.; Shih, J.; Brunet, A.; Rando, T.A. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013, 4, 189–204. [Google Scholar] [CrossRef] [Green Version]
- Sreepadmanabh, M.; Toley, B.J. Investigations into the cancer stem cell niche using in-vitro 3-D tumor models and microfluidics. Biotechnol. Adv. 2018, 36, 1094–1110. [Google Scholar] [CrossRef]
- LaBarbera, D.V.; Reid, B.G.; Yoo, B.H. The multicellular tumor spheroid model for high-throughput cancer drug discovery. Expert Opin. Drug. Discov. 2012, 7, 819–883. [Google Scholar] [CrossRef]
- Forde, S.; Matthews, J.D.; Jahangiri, L.; Lee, L.C.; Prokoph, N.; Malcolm, T.I.; Giger, O.T.; Bell, N.; Blair, H.; O’Marcaigh, A.; et al. Paediatric Burkitt lymphoma patient-derived xenografts capture disease characteristics over time and are a model for therapy. Br. J. Haematol. 2021, 192, 354–365. [Google Scholar] [CrossRef]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, F.; Gopalan, V.; Wahab, R.; Smith, R.A.; Lam, A.K. Cancer stem cells in oesophageal squamous cell carcinoma: Identification, prognostic and treatment perspectives. Crit. Rev. Oncol. Hematol. 2015, 96, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.H.; Dai, Y.D.; Tong, M.; Chan, Y.P.; Kwanm, P.S.; Fu, L.; Qin, Y.R.; Tsao, S.W.; Lung, H.L.; Lung, M.L.; et al. A CD90(+) tumor-initiating cell population with an aggressive signature and metastatic capacity in esophageal Cancer. Cancer Res. 2013, 73, 2322–2332. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.C.; Chou, K.T.; Hsu, J.W.; Lin, J.H.; Hsu, T.W.; Yen, D.H.; Hung, S.C.; Hsu, H.S. High metabolic rate and stem cell characteristics of esophageal cancer stem-like cells depend on the Hsp27-AKT-HK2 pathway. Int. J. Cancer 2019, 145, 2144–2156. [Google Scholar] [CrossRef]
- Taniguchi, H.; Moriya, C.; Igarashi, H.; Saitoh, A.; Yamamoto, H.; Adachi, Y.; Imai, K. Cancer stem cells in human gastrointestinal Cancer. Cancer Sci. 2016, 107, 1556–1562. [Google Scholar] [CrossRef]
- Munro, M.J.; Wickremesekera, S.K.; Peng, L.; Tan, S.T.; Itinteang, T. Cancer stem cells in colorectal cancer: A review. J. Clin. Pathol. 2018, 71, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Clark, P.A.; Iida, M.; Treisman, D.M.; Kalluri, H.; Ezhilan, S.; Zorniak, M.; Wheeler, D.L.; Kuo, J.S. Activation of multiple ERBB family receptors mediates glioblastoma cancer stem-like cell resistance to EGFR-targeted inhibition. Neoplasia 2012, 14, 420–428. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.W.; Choi, S.W.; Yang, S.R.; Shin, T.H.; Kim, H.S.; Yu, K.R.; Hong, I.S.; Ro, S.; Cho, J.M.; Kang, K.S. Growth arrest and forced differentiation of human primary glioblastoma multiforme by a novel small molecule. Sci. Rep. 2014, 4, 5546. [Google Scholar] [CrossRef]
- Cheray, M.; Bessette, B.; Lacroix, A.; Mélin, C.; Jawhari, S.; Pinet, S.; Deluche, E.; Clavère, P.; Durand, K.; Sanchez-Prieto, R.; et al. KLRC3, a Natural Killer receptor gene, is a key factor involved in glioblastoma tumourigenesis and aggressiveness. J. Cell Mol. Med. 2017, 21, 244–253. [Google Scholar] [CrossRef]
- Wang, H.H.; Liao, C.C.; Chow, N.H.; Huang, L.L.; Chuang, J.I.; Wei, K.C.; Shin, J.W. Whether CD44 is an applicable marker for glioma stem cells. Am. J. Transl. Res. 2017, 9, 4785–4806. [Google Scholar] [PubMed]
- Yang, M.; Yan, M.; Zhang, R.; Li, J.; Luo, Z. Side population cells isolated from human osteosarcoma are enriched with tumor-initiating cells. Cancer Sci. 2011, 102, 1774. [Google Scholar] [CrossRef]
- Maurizi, G.; Verma, N.; Gadi, A.; Mansukhani, A.; Basilico, C. Sox2 is required for tumor development and cancer cell proliferation in osteosarcoma. Oncogene August 2018, 37, 4626–4632. [Google Scholar] [CrossRef] [PubMed]
- Deter, R.L.; De Duve, C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 1967, 33, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wang, X. The interplay between autophagy and the ubiquitin-proteasome system in cardiac proteotoxicity. Biochim. Biophys. Acta 2015, 1852, 188–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilienbaum, A. Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol. 2013, 4, 1–26. [Google Scholar] [PubMed]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mari, M.; Tooze, S.A.; Reggiori, F. The puzzling origin of the autophagosomal membrane. F1000 Biol. Rep. 2011, 3, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Cuervo, A.M.; Seglen, P.O. Methods for monitoring autophagy from yeast to human. Autophagy 2007, 3, 181–206. [Google Scholar] [CrossRef] [Green Version]
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell Biol. 2010, 12, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.; Ktistakis, N.T. Omegasomes: PI3P platforms that manufacture autophagosomes. Essays Biochem. 2013, 55, 17–27. [Google Scholar] [PubMed]
- Esclatine, A.; Chaumorcel, M.; Codogno, P. Macroautophagy signaling and regulation. Curr. Top. Microbiol. Immunol. 2009, 335, 33–70. [Google Scholar] [PubMed]
- Eskelinen, E.L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 2009, 1793, 664–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monastyrska, I.; Rieter, E.; Klionsky, D.J.; Reggiori, F. Multiple roles of the cytoskeleton in autophagy. Biol. Rev. Camb. Philos. Soc. 2009, 84, 431–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.G.; Macleod, K.F. Autophagy, cancer stem cells and drug resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praharaj, P.P.; Panigrahi, D.P.; Bhol, C.S.; Patra, S.; Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Singh, A.; Patil, S.; Bhutia, S.K. Mitochondrial rewiring through mitophagy and mitochondrial biogenesis in cancer stem cells: A potential target for anti-CSC cancer therapy. Cancer Lett. 2021, 498, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Van Doeselaar, S.; Burgering, B.M.T. FOXOs Maintaining the Equilibrium for Better or for Worse. Curr. Top. Dev. Biol. 2018, 127, 49–103. [Google Scholar]
- Barzilay, R.; Melamed, E.; Offen, D. Introducing Transcription Factors to Multipotent Mesenchymal Stem Cells: Making Transdifferentiation Possible. Stem Cells 2009, 27, 2509–2515. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Qin, J.; Zhang, Y.; Cheng, X.; Wang, X.; Lu, W.; Xie, X.; Zhang, S. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J. Exp. Clin. Cancer Res. 2017, 36, 171. [Google Scholar] [CrossRef] [Green Version]
- Cufí, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Vellon, L.; Menendez, J.A. Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle. 2011, 10, 3871–3885. [Google Scholar] [CrossRef] [Green Version]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Kiyono, K.; Suzuki, H.I.; Matsuyama, H.; Morishita, Y.; Komuro, A.; Kano, M.R.; Sugimoto, K.; Miyazono, K. Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 2009, 69, 8844–8852. [Google Scholar] [CrossRef] [Green Version]
- Morel, A.P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef]
- Courtois, S.; Haykal, M.; Bodineau, C.; Sifré, E.; Azzi-Martin, L.; Ménard, A.; Mégraud, F.; Lehours, P.; Durán, R.V.; Varon, C.; et al. Autophagy induced by Helicobacter pylori infection is necessary for gastric cancer stem cell emergence. Gastric. Cancer 2021, 24, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Flynn, A.B.; Schiemann, W.P. Autophagy in breast cancer metastatic dormancy: Tumor suppressing or tumor promoting functions? J. Cancer Metastasis Treat. 2019, 5, 43. [Google Scholar] [PubMed]
- Chaterjee, M.; van Golen, K.L. Breast cancer stem cells survive periods of farnesyl-transferase inhibitor-induced dormancy by undergoing autophagy. Bone Marrow Res. 2011, 2011, 362938. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Deloménie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef]
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 2015, 13, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Ojha, R.; Bhattacharyya, S.; Singh, S.K. Autophagy in Cancer Stem Cells: A Potential Link Between Chemoresistance, Recurrence, and Metastasis. Biores. Open Access. 2015, 4, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Mele, L.; del Vecchio, V.; Liccardo, D.; Prisco, C.; Schwerdtfeger, M.; Robinson, N.; Desiderio, V.; Tirino, V.; Papaccio, G.; La Noce, M. The role of autophagy in resistance to targeted therapies. Cancer Treat Rev. 2020, 88, 102043. [Google Scholar] [CrossRef]
- Golden, E.B.; Cho, H.Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus. 2014, 37, E12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.-Q.; Pan, D.; Zhang, S.-W.; Xie, D.-Y.; Zheng, X.-L.; Chen, H. Autophagy regulates chemoresistance of gastric cancer stem cells via the Notch signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3402–3407. [Google Scholar]
- Fu, Y.; Chang, H.; Peng, X.; Bai, Q.; Yi, L.; Zhou, Y.; Zhu, J.; Mi, M. Resveratrol inhibits breast cancer stem-like cells and induces autophagy via suppressing Wnt/β-catenin signaling pathway. PLoS ONE 2014, 9, e102535. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.A.; Saha, S.K.; Rahman, M.S.; Uddin, M.J.; Uddin, M.S.; Pang, M.-G.; Rhim, H.; Cho, S.G. Molecular Insights Into Therapeutic Potential of Autophagy Modulation by Natural Products for Cancer Stem Cells. Front. Cell Dev. Biol. 2020, 8, 283. [Google Scholar] [CrossRef]
- Mandhair, H.K.; Arambasic, M.; Novak, U.; Radpour, R. Molecular modulation of autophagy: New venture to target resistant cancer stem cells. World J. Stem Cells. 2020, 12, 303–322. [Google Scholar] [CrossRef] [PubMed]
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C.; et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2019, 33, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Terry, S.; Faouzi Zaarour, R.; Hassan Venkatesh, G.; Francis, A.; El-Sayed, W.; Buart, S.; Bravo, P.; Thiery, J.; Chouaib, S. Role of Hypoxic Stress in Regulating Tumor Immunogenicity, Resistance and Plasticity. Int. J. Mol. Sci. 2018, 19, 3044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, W.; Tong, G.; Liu, Y. Cancer stem cells and hypoxia-inducible factors (Review). Int. J. Oncol. 2018, 53, 469–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Dynamic regulation of stem cell specification and maintenance by hypoxia-inducible factors. Mol. Aspects Med. 2016, 47, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Wu, M.; Chiou, S.; Chen, P.; Chang, S.; Liu, C.; Teng, S.; Wu, K. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Nakuluri, K.; Mukhi, D.; Nishad, R.; Saleem, M.A.; Mungamuri, S.K.; Menon, R.K.; Pasupulati, A.K. Hypoxia induces ZEB2 in podocytes: Implications in the pathogenesis of proteinuria. J. Cell Physiol. 2019, 234, 6503–6518. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Tan, X.; Tampe, B.; Sanchez, E.; Zeisberg, M.; Zeisberg, E.M. ducible Factor 1α (HIF1α) in Hypoxia-induced Endothelial to Mesenchymal Transition of Human Coronary Endothelial Cells. J. Biol. Chem. 2015, 290, 16653–16664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Hasmim, M.; Janji, B.; Khaled, M.; Noman, M.Z.; Louache, F.; Bordereaux, D.; Abderamane, A.; Baud, V.; Mami-Chouaib, F.; Chouaib, S. Cutting Edge: NANOG Activates Autophagy under Hypoxic Stress by Binding to BNIP3L Promoter. J. Immunol. 2017, 198, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, D.; Liu, Y.; Su, Z.; Zhang, L.; Chen, F.; Zhou, Y.; Wu, Y.; Yu, M.; Zhang, Z.; et al. Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell Int. 2013, 13, 119. [Google Scholar] [CrossRef] [Green Version]
- Qureshi-Baig, K.; Kuhn, D.; Viry, E.; Pozdeev, V.I.; Schmitz, M.; Rodriguez, F.; Ullmann, P.; Koncina, E.; Nurmik, M.; Frasquilho, S.; et al. Hypoxia-induced autophagy drives colorectal cancer initiation and progression by activating the PRKC/PKC-EZR (ezrin) pathway. Autophagy 2020, 16, 1436–1452. [Google Scholar] [CrossRef] [Green Version]
- Yuan, N.; Song, L.; Zhang, S.; Lin, W.; Cao, Y.; Xu, F.; Fang, Y.; Wang, Z.; Zhang, H.; Li, X.; et al. Bafilomycin A1 targets both autophagy and apoptosis pathways in pediatric B-cell acute lymphoblastic leukemia. Haematologica 2015, 100, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Wouters, B.; Koritzinsky, M. Hypoxia signalling through mTOR and the unfolded protein response in Cancer. Nat. Rev. Cancer 2008, 8, 851–864. [Google Scholar] [CrossRef]
- Land, S.C.; Tee, A.R. Hypoxia-inducible Factor 1α Is Regulated by the Mammalian Target of Rapamycin (mTOR) via an mTOR Signaling Motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agani, F.; Jiang, B.H. Oxygen-independent Regulation of HIF-1: Novel Involvement of PI3K/ AKT/mTOR Pathway in Cancer Bd. 13. Curr. Cancer Drug Targets 2013, 3, 245–251. [Google Scholar] [CrossRef]
- Losuwannarak, N.; Maiuthed, A.; Kitkumthorn, N.; Leelahavanichkul, A.; Roytrakul, S.; Chanvorachote, P. Gigantol Targets Cancer Stem Cells and Destabilizes Tumors via the Suppression of the PI3K/AKT and JAK/STAT Pathways in Ectopic Lung Cancer Xenografts. Cancers 2019, 11, 2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Liu, X.; Xu, Y.; Liu, J.; Xie, M.; Ni, W.; Chen, S. KLF5 promotes hypoxia-induced survival and inhibits apoptosis in non-small cell lung cancer cells via HIF-1α. Int. J. Oncol. 2014, 45, 1507–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, T.; Cui, L.; Wang, H.; Wang, H.; Han, N. Knockdown of KLF5 suppresses hypoxia-induced resistance to cisplatin in NSCLC cells by regulating HIF-1α-dependent glycolysis through inactivation of the PI3K/Akt/mTOR pathway. J. Transl. Med. 2018, 16, 164. [Google Scholar] [CrossRef] [Green Version]
- Nazio, F.; Bordi, M.; Cianfanelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Peixoto, J.; Lima, J. Metabolic traits of cancer stem cells. Dis. Model. Mech. 2018, 11, dmm033464. [Google Scholar] [CrossRef] [Green Version]
- Lleonart, M.E.; Abad, E.; Graifer, D.; Lyakhovich, A. Reactive Oxygen Species-Mediated Autophagy Defines the Fate of Cancer Stem Cells. Antioxid. Redox Signal. 2018, 28, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Held, N.M.; Houtkooper, R.H. Mitochondrial quality control pathways as determinants of metabolic health. Bioessays 2015, 37, 867–876. [Google Scholar] [CrossRef]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. PNAS 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Wu, Q.; Horbinski, C.M.; Flavahan, W.A.; Yang, K.; Zhou, W.; Dombrowski, S.M.; Huang, Z.; Fang, X.; Shi, Y.; et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat. Neurosci. 2015, 18, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell. 2017, 68, 281–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Luo, L.; Guo, C.-Y.; Goto, S.; Urata, Y.; Shao, J.-H.; Li, T.-S. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017, 388, 34–42. [Google Scholar] [CrossRef]
- Yan, C.; Li, T.S. Dual Role of Mitophagy in Cancer Drug Resistance. Anticancer Res. 2018, 38, 617–621. [Google Scholar] [PubMed] [Green Version]
- Zhou, J.; Li, G.; Zheng, Y.; Shen, H.M.; Hu, X.; Ming, Q.L.; Huang, C.; Li, P.; Gao, N. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 2015, 11, 1259–1279. [Google Scholar] [CrossRef] [Green Version]
- Pennisi, E. Genomics. ENCODE project writes eulogy for junk DNA. Science 2012, 337, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Ponjavic, J.; Ponting, C.P.; Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007, 17, 556–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Liu, C.; Zhao, Q.; Lü, J.; Ding, X.; Luo, A.; He, J.; Wang, G.; Li, Y.; Cai, Z.; et al. Long non-coding RNA CCAT2 promotes oncogenesis in triple-negative breast cancer by regulating stemness of cancer cells. Pharmacol. Res. 2020, 152, 104628. [Google Scholar] [CrossRef]
- Wu, J.; Zhu, P.; Lu, T.; Du, Y.; Wang, Y.; He, L.; Ye, B.; Liu, B.; Yang, L.; Wang, J.; et al. The long non-coding RNA LncHDAC2 drives the self-renewal of liver cancer stem cells via activation of Hedgehog signaling. J. Hepatol. 2019, 70, 918–929. [Google Scholar] [CrossRef]
- Crea, F.; Clermont, P.L.; Parolia, A.; Wang, Y.; Helgason, C.D. The non-coding transcriptome as a dynamic regulator of cancer metastasis. Cancer Metastasis Rev. 2014, 33, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crea, F.; Venalainen, E.; Ci, X.; Cheng, H.; Pikor, L.; Parolia, A.; Xue, H.; Nur Saidy, N.R.; Lin, D.; Lam, W.; et al. The role of epigenetics and long noncoding RNA MIAT in neuroendocrine prostate. Cancer Epigenomics 2016, 8, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, F.; Hu, H.; Han, T.; Yuan, C.; Wang, L.; Jin, Z.; Guo, Z.; Wang, L. Long noncoding RNA MALAT-1 enhances stem cell-like phenotypes in pancreatic cancer cells. Int. J. Mol. Sci. 2015, 16, 6677–6693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peperstraete, E.; Lecerf, C.; Collette, J.; Vennin, C.; Raby, L.; Völkel, P.; Angrand, P.O.; Winter, M.; Bertucci, F.; Finetti, P.; et al. Enhancement of Breast Cancer Cell Aggressiveness by lncRNA H19 and its Mir-675 Derivative: Insight into Shared and Different Actions. Cancers 2020, 29, 1730. [Google Scholar] [CrossRef]
- Dai, D.; Wang, H.; Zhu, L.; Jin, H.; Wang, X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- Siebenthall, K.T.; Miller, C.P.; Vierstra, J.D.; Mathieu, J.; Tretiakova, M.; Reynolds, A.; Sandstrom, R.; Rynes, E.; Haugen, E.; Johnson, A.; et al. Integrated epigenomic profiling reveals endogenous retrovirus reactivation in renal cell carcinoma. EBioMedicine 2019, 41, 427–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Bu, P. Non-coding RNAs in cancer stem cells. Cancer Lett. 2018, 421, 121–126. [Google Scholar] [CrossRef]
- Huang, R.; Zhu, L.; Zhang, Y. XIST lost induces ovarian cancer stem cells to acquire taxol resistance via a KMT2C-dependent way. Cancer Cell Int. 2020, 20, 436. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Xu, J.; Lin, Z.; Lu, Y.; Xin, X.; Li, X.; Yang, Y.; Meng, Q.; Wang, C.; Xiong, W.; et al. Inflammatory factor receptor Toll-like receptor 4 controls telomeres through heterochromatin protein 1 isoforms in liver cancer stem cell. J. Cell Mol. Med. 2018, 22, 3246–3258. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; An, J.; Wu, M.; Zheng, Q.; Gui, X.; Li, T.; Pu, H.; Lu, D. LncRNA HOTAIR promotes human liver cancer stem cell malignant growth through downregulation of SETD2. Oncotarget 2015, 6, 27847–27864. [Google Scholar] [CrossRef]
- Li, L.; Dang, Q.; Xie, H.; Yang, Z.; He, D.; Liang, L.; Song, W.; Yeh, S.; Chang, C. Correction: Infiltrating mast cells enhance prostate cancer invasion via altering LncRNA-HOTAIR/PRC2-androgen receptor (AR)-MMP9 signals and increased stem/progenitor cell population. Oncotarget 2016, 7, 83828. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, N.; Toyoda, M.; Yoshimura, H.; Matsuda, Y.; Arai, T.; Takubo, K.; Aida, J.; Ishiwata, T. H19 long non-coding RNA contributes to sphere formation and invasion through regulation of CD24 and integrin expression in pancreatic cancer cells. Oncotarget 2018, 9, 34719–34734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shima, H.; Kida, K.; Adachi, S.; Yamada, A.; Sugae, S.; Narui, K.; Miyagi, Y.; Nishi, M.; Ryo, A.; Murata, S.; et al. Lnc RNA H19 is associated with poor prognosis in breast cancer patients and promotes cancer stemness. Breast Cancer Res. Treat. 2018, 170, 507–516. [Google Scholar] [CrossRef]
- Zhan, Y.; Chen, Z.; He, S.; Gong, Y.; He, A.; Li, Y.; Zhang, L.; Zhang, X.; Fang, D.; Li, X.; et al. Long non-coding RNA SOX2OT promotes the stemness phenotype of bladder cancer cells by modulating SOX2. Mol. Cancer 2020, 19, 25. [Google Scholar] [CrossRef]
- Zheng, A.; Song, X.; Zhang, L.; Zhao, L.; Mao, X.; Wei, M.; Jin, F. Long non-coding RNA LUCAT1/miR-5582-3p/TCF7L2 axis regulates breast cancer stemness via Wnt/β-catenin pathway. J. Exp. Clin. Cancer Res. 2019, 38, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucci, P.; Rescigno, P.; Sumanasuriya, S.; de Bono, J.; Crea, F. Hypoxia and Noncoding RNAs in Taxane Resistance. Trends Pharmacol. Sci. 2018, 39, 695–709. [Google Scholar] [CrossRef]
- Wang, X.; Sun, W.; Shen, W.; Xia, M.; Chen, C.; Xiang, D.; Ning, B.; Cui, X.; Li, H.; Li, X.; et al. Long non-coding RNA DILC regulates liver cancer stem cells via IL-6/STAT3 axis. J. Hepatol. 2016, 64, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Nie, W.; Yao, K.; Chou, J. Depletion of the lncRNA RP11-567G11.1 inhibits pancreatic cancer progression. Biomed. Pharmacother. 2019, 112, 108685. [Google Scholar] [CrossRef] [PubMed]
- Pucci, P.; Venalainen, E.; Alborelli, I.; Quagliata, L.; Hawkes, C.; Mather, R.; Romero, I.; Rigas, S.H.; Wang, Y.; Crea, F. LncRNA HORAS5 promotes taxane resistance in castration-resistant prostate cancer via a BCL2A1-dependent mechanism. Epigenomics 2020, 12, 1123–1138. [Google Scholar] [CrossRef]
- Qiu, G.; Ma, D.; Li, F.; Sun, D.; Zeng, Z. lnc-PKD2-2-3, identified by long non-coding RNA expression profiling, is associated with pejorative tumor features and poor prognosis, enhances cancer stemness and may serve as cancer stem-cell marker in cholangiocarcinoma. Int. J. Oncol. 2019, 55, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellmunt, J.; Eigl, B.J.; Senkus, E.; Loriot, Y.; Twardowski, P.; Castellano, D.; Blais, N.; Sridhar, S.S.; Sternberg, C.N.; Retz, M.; et al. Borealis-1: A randomized, first-line, placebo-controlled, phase II study evaluating apatorsen and chemotherapy for patients with advanced urothelial Cancer. Ann. Oncol. 2017, 28, 2481–2488. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Higano, C.S.; Blumenstein, B.; Ferrero, J.M.; Reeves, J.; Feyerabend, S.; Gravis, G.; Merseburger, A.S.; Stenzl, A.; Bergman, A.M.; et al. Custirsen in combination with docetaxel and prednisone for patients with metastatic castration-resistant prostate cancer (SYNERGY trial): A phase 3, multicentre, open-label, randomised trial. Lancet Oncol. 2017, 18, 473–485. [Google Scholar] [CrossRef]
- Chery, J. RNA therapeutics: RNAi and antisense mechanisms and clinical applications. Postdoc J. 2016, 4, 35–50. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.T.; Han, C.; Sun, Y.M.; Chen, T.Q.; Chen, Y.Q. Noncoding RNAs in cancer therapy resistance and targeted drug development. J. Hematol. Oncol. 2019, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, H.S.; London, C.A.; Sekhon, M.; Iversen, P.L.; Devi, G.R. c-MYC antisense phosphosphorodiamidate morpholino oligomer inhibits lung metastasis in a murine tumor model. Lung Cancer 2008, 60, 347–354. [Google Scholar] [CrossRef]
- Iversen, P.L.; Arora, V.; Acker, A.J.; Mason, D.H.; Devi, G.R. Efficacy of antisense morpholino oligomer targeted to c-myc in prostate cancer xenograft murine model and a Phase I safety study in humans. Clin. Cancer Res. 2003, 9, 2510–2519. [Google Scholar]
- Vidovic, D.; Huynh, T.T.; Konda, P.; Dean, C.; Cruickshank, B.M.; Sultan, M.; Coyle, K.M.; Gujar, S.; Marcato, P. ALDH1A3-regulated long non-coding RNA NRAD1 is a potential novel target for triple-negative breast tumors and cancer stem cells. Cell Death Differ. 2020, 27, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Lu, H.Y.; Xia, Y.H.; Jiang, A.G.; Lv, Y.X. Long non-coding RNA EPIC1 promotes human lung cancer cell growth. Biochem. Biophys. Res. Commun. 2018, 503, 1342–1348. [Google Scholar] [CrossRef]
- Panda, S.; Setia, M.; Kaur, N.; Shepal, V.; Arora, V.; Singh, D.K.; Mondal, A.; Teli, A.; Tathode, M.; Gajula, R.; et al. Noncoding RNA Ginir functions as an oncogene by associating with centrosomal proteins. PLoS Biol. 2018, 16, e2004204. [Google Scholar] [CrossRef] [Green Version]
- Lavalou, P.; Eckert, H.; Damy, L.; Constanty, F.; Majello, S.; Bitetti, A.; Graindorge, A.; Shkumatava, A. Corrigendum: Strategies for genetic inactivation of long noncoding RNAs in zebrafish. RNA 2020, 26, 529. [Google Scholar] [CrossRef] [Green Version]
- Roobol, M.J.; Schröder, F.H.; van Leeuwen, P.; Wolters, T.; van den Bergh, R.C.; van Leenders, G.J.; Hessels, D. Performance of the prostate cancer antigen 3 (PCA3) gene and prostate-specific antigen in prescreened men: Exploring the value of PCA3 for a first-line diagnostic test. Eur. Urol. 2010, 58, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Su, Y.; Liu, X.; Xu, M.; Chen, X.; Zhu, Y.; Guo, Z.; Bai, T.; Dong, L.; Wei, C.; et al. Serum and exosome long non coding RNAs as potential biomarkers for hepatocellular carcinoma. J. Cancer 2018, 9, 2631–2639. [Google Scholar] [CrossRef]
- Conigliaro, A.; Costa, V.; Lo Dico, A.; Saieva, L.; Buccheri, S.; Dieli, F.; Manno, M.; Raccosta, S.; Mancone, C.; Tripodi, M.; et al. CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19, l.n.c.R.N.A. Mol. Cancer 2015, 14, 155. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Chen, C.; Zhou, Q.; Wang, Y.; Zhao, Y.; Zhao, X.; Li, W.; Zheng, S.; Ye, H.; Wang, L.; et al. LncRNA HOTTIP modulates cancer stem cell properties in human pancreatic cancer by regulating HOXA9. Cancer Lett. 2017, 410, 68–81. [Google Scholar] [CrossRef]
- Han, M.; Gu, Y.; Lu, P.; Li, J.; Cao, H.; Li, X.; Qian, X.; Yu, C.; Yang, Y.; Yang, X.; et al. Exosome-mediated lncRNA AFAP1-AS1 promotes trastuzumab resistance through binding with AUF1 and activating ERBB2 translation. Mol. Cancer 2020, 19, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Wang, H.; Pang, Y.; Hu, H.; Zhang, H.; Wang, W. Exosomal long non-coding RNA UCA1 functions as growth inhibitor in esophageal Cancer. Aging 2020, 12, 20523–20539. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Dawson, E.; Duong, A.; Haw, R.; Stein, L. ReactomeFIViz: A Cytoscape app for pathway and network-based data analysis. F1000Research 2014, 3, 146. [Google Scholar]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Piñero, J.; Ramírez-Anguita, J.M. Saüch-Pitarch J, Ronzano F, Centeno E, Sanz F, Furlong LI. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2019, 48, D845–D885. [Google Scholar]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMI J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Montojo, J.; Zuberi, K.; Rodriguez, H.; Kazi, F.; Wright, G.; Donaldson, S.L.; Morris, Q.; Bader, G.D. GeneMANIA Cytoscape plugin: Fast gene function predictions on the desktop. Bioinformatics 2010, 26, 2927–2928. [Google Scholar] [CrossRef]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment Map: A Network-Based Method for Gene-Set Enrichment Visualization and Interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef]
- Terry, S.; Buart, S.; Tan, T.Z.; Gros, G.; Noman, M.Z.; Lorens, J.B.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. Acquisitionoftumor cell phenotypic diversity along the EMT spectrum under hypoxic pressure: Consequences on susceptibility to cell-mediated cytotoxicity. Oncoimmunology 2017, 6, e1271858. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Dong, B.; Cao, J.; Mao, Y.; Guan, W.; Peng, Y.; Wang, S. Long non-coding RNA in glioma: Signaling pathways. Oncotarget 2017, 8, 27582–27592. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.S.; Jiang, J.; Liang, X.H.; Tang, Y.L. Links between cancer stem cells and epithelial-mesenchymal transition. Onco. Targets Ther. 2015, 8, 2973–2980. [Google Scholar] [PubMed] [Green Version]
- Galoczova, M.; Coates, P.; Vojtesek, B. STAT3, stem cells, cancer stem cells and p63. Cell Mol. Biol. Lett. 2018, 23, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Zhao, J.; Qin, Y.; Zhao, F.; Ji, L.; Zhang, J. Overexpression of ATG4a promotes autophagy and proliferation, and inhibits apoptosis in lens epithelial cells via the AMPK and Akt pathways. Mol. Med. Rep. 2020, 22, 1295–1302. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Lin, Z.; Flomenberg, N.; Howell, A.; Pestell, R.G.; Lisanti, M.P.; Sotgia, F. Cytokine production and inflammation drive autophagy in the tumor microenvironment: Role of stromal caveolin-1 as a key regulator. Cell Cycle. 2011, 10, 1784–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visvader, J.E. Cells of origin in Cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- Taussig, D.C.; Miraki-Moud, F.; Anjos-Afonso, F.; Pearce, D.J.; Allen, K.; Ridler, C.; Lillington, D.; Oakervee, H.; Cavenagh, J.; Agrawal, S.G. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood 2008, 112, 568–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.E.; Eom, J.I.; Jeung, H.K.; Cheong, J.W.; Lee, J.Y.; Kim, J.S.; Min, Y.H. Targeting AMPK-ULK1-mediated autophagy for combating BET inhibitor resistance in acute myeloid leukemia stem cells. Autophagy 2017, 13, 761–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Nguyen, L.X.T.; Li, L.; Zhao, D.; Kumar, B.; Wu, H.; Lin, A.; Pellicano, F.; Hopcroft, L.; Su, Y.-L.; et al. Bone marrow niche trafficking of miR-126 controls the self-renewal of leukemia stem cells in chronic myelogenous leukemia. Nat. Med. 2018, 24, 450–462. [Google Scholar] [CrossRef]
- Silvestri, G.; Trotta, R.; Stramucci, L.; Ellis, J.J.; Harb, J.G.; Neviani, P.; Wang, S.; Eisfeld, A.K.; Walker, C.J.; Zhang, B.; et al. Persistence of Drug-Resistant Leukemic Stem Cells and Impaired NK Cell Immunity in CML Patients Depend on MIR300 Antiproliferative and PP2A-Activating Functions. Blood. Cancer Discov. 2020, 1, 48–67. [Google Scholar] [CrossRef]
- Wang, L.; Bu, P.; Ai, Y.; Srinivasan, T.; Chen, H.J.; Xiang, K.; Lipkin, S.M.; Shen, X. A long non-coding RNA targets microRNA miR-34a to regulate colon cancer stem cell asymmetric division. Elife 2016, 5, e14620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhu, L.; Xu, L.; Qin, K.; Liu, C.; Yu, Y.; Su, D.; Wu, K.; Sheng, Y. Long noncoding RNA linc00617 exhibits oncogenic activity in breast Cancer. Mol. Carcinog. 2017, 56, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Notta, F.; Mullighan, C.G.; Wang, J.C.; Poeppl, A.; Doulatov, S.; Phillips, L.A.; Ma, J.; Minden, M.D.; Downing, J.R.; Dick, J.E. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 2011, 469, 362–367. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Examples of Surface Markers, Stemness Proteins, or Factors Supporting the Maintenance of Stemness |
|---|---|
| AML | CD34+ CD38- or CD34- cells [17,28,29,30] |
| Oesophageal cancer | B7H4, LETM1, CD90 [31,32] |
| Colorectal cancer | CD44, CD133, ST6GALNAC1 [33] |
| Gastric cancer | CD44, SOX2, KLF4, and OCT4 [34,35] |
| Pancreatic cancer | CD133, CD24, CD44, ESA [36,37] |
| Liver cancer (hepatocellular carcinoma) | CD13, CD24, CD44, CD90, CD133 and EpCAM [38] |
| Lung Cancer (Non-small cell lung cancer) | SOX2 and NANOG [39] CDKN1A, SNAI1, and ITGA6 [40] |
| Glioblastoma multiforme | CD133 [41] SOX2, OCT4, NANOG, and SALL4 [42] |
| Osteosarcoma | SOX2 [43] |
| Breast cancer | CD44+ CD24-/low ALDH1+ [13,44] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahangiri, L.; Ishola, T.; Pucci, P.; Trigg, R.M.; Pereira, J.; Williams, J.A.; Cavanagh, M.L.; Gkoutos, G.V.; Tsaprouni, L.; Turner, S.D. The Role of Autophagy and lncRNAs in the Maintenance of Cancer Stem Cells. Cancers 2021, 13, 1239. https://doi.org/10.3390/cancers13061239
Jahangiri L, Ishola T, Pucci P, Trigg RM, Pereira J, Williams JA, Cavanagh ML, Gkoutos GV, Tsaprouni L, Turner SD. The Role of Autophagy and lncRNAs in the Maintenance of Cancer Stem Cells. Cancers. 2021; 13(6):1239. https://doi.org/10.3390/cancers13061239
Chicago/Turabian StyleJahangiri, Leila, Tala Ishola, Perla Pucci, Ricky M. Trigg, Joao Pereira, John A. Williams, Megan L. Cavanagh, Georgios V. Gkoutos, Loukia Tsaprouni, and Suzanne D. Turner. 2021. "The Role of Autophagy and lncRNAs in the Maintenance of Cancer Stem Cells" Cancers 13, no. 6: 1239. https://doi.org/10.3390/cancers13061239