A chromosome-level genome assembly of the Walking goby (Scartelaos histophorus)
 Xuanzhao Li
Xuanzhao Li Jiwei Qi1†
Jiwei Qi1†  Joel B. Corush
Joel B. Corush Jiajie Chen
Jiajie Chen Jie Zhang
Jie Zhang- 1Chinese Academy of Sciences Key Laboratory of Animal Ecology and Conservation Biology, Institute of Zoology, Beijing, China
- 2School of Life Sciences, Institute of Life Sciences and Green Development, Hebei University, Baoding, China
- 3Illinois Natural History Survey, Prairie Research Institute, University of Illinois Urbana-Champaign, Champaign, IL, United States
- 4Marine Life Science Museum, East China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Shanghai, China
Introduction
The moniker “Mudskipper” typically refers to four genera of the family Oxudercidae, including Scartelaos, Boleophthalmus, Periophthalmodon, and Periophthalmus (Patzner et al., 2011; Nelson et al., 2016). Mudskippers are amphibious teleosts that can be found on mudflats and mangroves across the Indo-Pacific and Southeastern Atlantic Ocean. They have many morphological and physiological traits that allow for their amphibious life history. Dorsally protruding eyes and other adaptive physiological traits provide clear vision in both air and water (Clayton, 1993; Sayer, 2005; Hu et al., 2022). Developed capillaries in the epidermis (Zhang et al., 2000; 2003) and specialized gill structure aid in respirations during prolonged out-of-water periods (Pan et al., 2010). Modified musculature and skeletal structures such as well-developed pectoral fins provide support for crawling or jumping on mudflats to feed and escape predation (Pace and Gibb, 2009; Wang et al., 2013; You et al., 2018). This terrestrial life history is also important in biogeographical patterns of these fishes (Corush et al., 2022). Together, these adaptive strategies make mudskippers ideal for studying evolutionary transitions between land and water.
Across these four genera, a decrease in time spent in aquatic environments occurs in the following order: Scartelaos > Boleophthalmus > Periophthalmodon > Periophthalmus (Zhang, 2001). These genera have different degrees of terrestriality resulting in variation in evolutionary pressures and ecological niches. Currently, only two Periophthalmus species have chromosome-scale genome data. In this study, we produced a high-quality chromosome-level reference genome of S. histophorus using PacBio CCS (circular consensus sequencing) and Hi-C (high-throughput chromosome conformation capture) technology. These genome data provide the necessary resource to address questions pertaining to water-to-land transition including aerial vision, terrestrial locomotion, and ammonia tolerance, with respect to not only the molecular mechanism but also evolutionary trajectories of chromosomes.
Data
We produced a high-quality chromosome-level genome of the Walking goby using PacBio CCS and Hi-C technology. Full DNA sequencing resulted in a total of 21.06 gigabases of PacBio HiFi reads, 99.88 gigabases of Hi-C reads, 50.64 gigabases Illumina reads generated. For RNA-seq, a total of 29.50 gigabases of Illumina reads were generated (Supplementary Table 1). Genome survey results show that the genome sizes of S. histophorus was ~817.12 Mb, with a heterozygosity ratio of ~0.62%, and the repeat sequence ratio was ~47.54% (Supplementary Figure 1; Supplementary Table 2). The final assembly genome size is 869.5 Mb, with contig N50 and scaffold N50 values of 9.02 and 35.28 Mb, respectively. A total of 616 contigs, which accounted for 91.42% (~794.9 Mb) of the total assembled genome, were anchored into 24 chromosomes (Figure 1; Supplementary Table 3). This is consistent with previous research (Arai, 2011). Based on BUSCO analysis, the final genome assembly included 96.7% and 97.4% of complete conserved orthologs within vertebrates and Actinopterygii, respectively (Supplementary Table 4). Illumina short reads were mapped onto the assembled genome, and a total of 99.05% of the reads were mapped covering 99.87% in the genome (Supplementary Table 5).
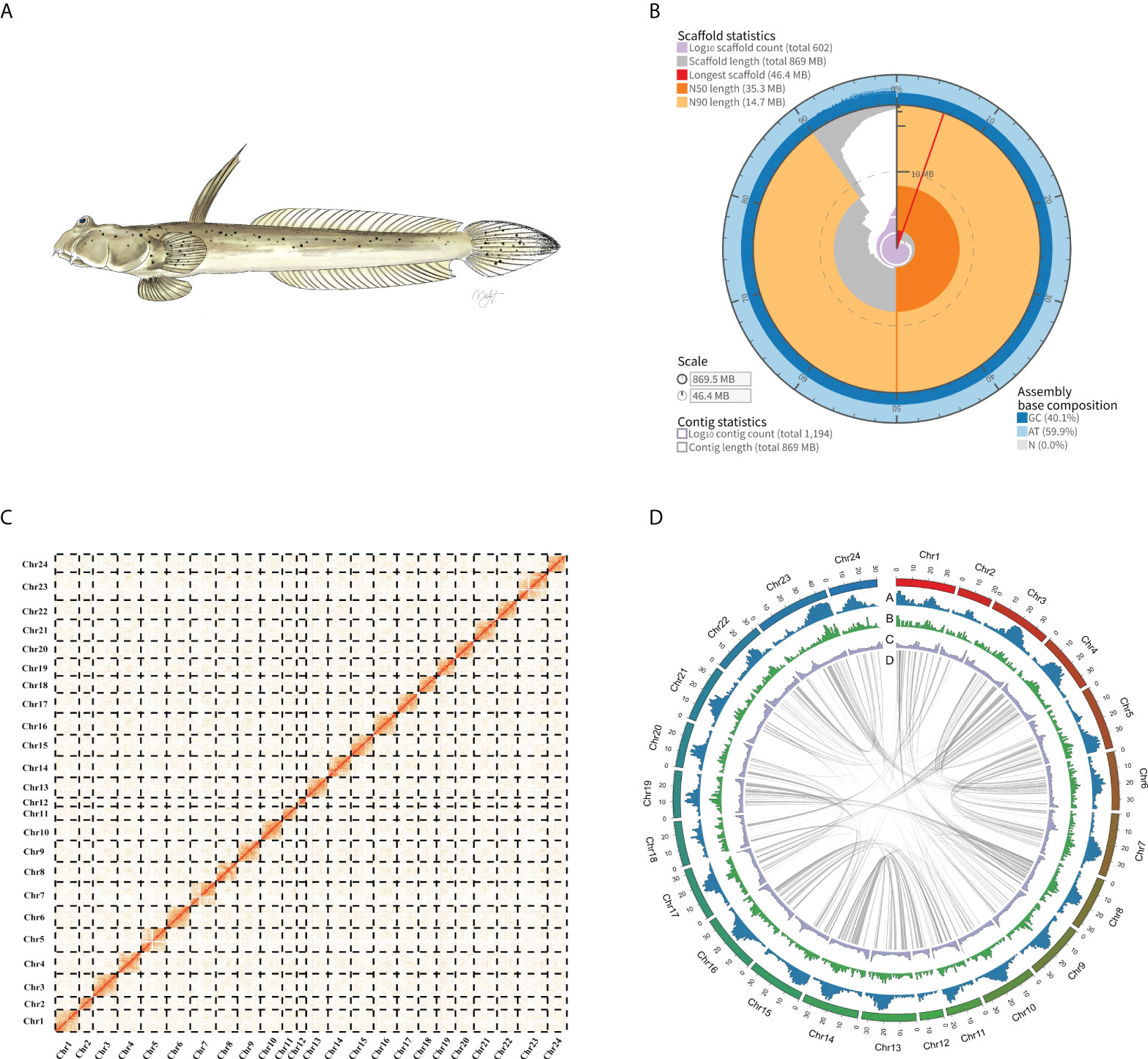
Figure 1 (A) A picture of Walking goby (Scartelaos histophorus). (B) Assembly metric visualizations (https://github.com/rjchallis/assembly-stats): The inner radius (red) represents the longest scaffold in the assembly genome. The arcs that connect to the radial axis indicate N50 (dark orange) and N90 scaffold (light orange) lengths. Segments plotted from the circumference (gray) and the length of segment at a given percentage indicate the cumulative percentage of the assembly that is contained within scaffolds of at least that length; the angle indicates the percentage of the assembly. The cumulative number of scaffolds within a given percentage of the genome (purple) originates at the center of the plot. (C) Hi-C matrix of the S. histophorus genome. For a more detailed, within-chromosome visualization, see Supplementary Figure 2. (D) Circos plot of S. histophorus genome. Numbers along the outermost circle represent number of base pairs in millions. (A) GC content, (B) gene density, (C) Repeat sequence density; all statistics use the 100-kb window. (D) Collinear gene blocks in the genome.
Repetitive sequences were annotated using both homology-based search and de novo approaches. The integrated results indicated that the S. histophorus genome contains 49.07% repetitive sequences (Supplementary Table 6). A total of 24,223 protein-coding genes were predicted and annotated, with an average exon number per gene of 9.59 and an average CDS length of 1,613.28 bp (Supplementary Table 7). BUSCO evaluation shows that the annotated protein-coding genes in the genome covered 90.9% of complete conserved ortholog genes within the vertebrate database and 90.7% of complete conserved ortholog genes within the Actinopterygii database (Supplementary Table 8), and 93.72% of these genes could be annotated by at least one of public database (NR, GO, KEGG, Swiss-Prot databases) (Supplementary Table 9).
The genome of S. histophorus was first reported in 2014 (You et al., 2014); however, this genome used whole-genome shotgun sequencing, and did not anchor reads to chromosomes, limiting its use in further mudskipper genomic research. By using three different methods, namely, shotgun short reads, PacBio CCS reads, and Hi-C technology, the new whole-genome quality was improved significantly. The contig N50 and scaffold N50 values of the previous genome are 8 and 14 kb, respectively. Using the Hi-C technology in our study, the contig N50 and scaffold N50 values of the genome were increased to 9.02 and 35.28 Mb, respectively. Furthermore, only 74.3% (vertebrates database) and 75.5% (Actinopterygii database) of the orthologs were detected in the previous genome from You et al. (2014), whereas our new S. histophorus genome includes 96.7% and 97.4% of complete conserved ortholog genes within the vertebrate database and Actinopterygii database. BUSCO analysis also showed that more protein-coding genes were predicted in our work and the chromosome-level genome continuity and completeness have improved compared to the previous study (Supplementary Table 8).
We used 4,280 single-copy homologous genes of Scartelaos histophorus, and 19 other species were used to construct a phylogenetic tree (Supplementary Table 10). All Gobiiformes (Scartelaos histophorus, Boleophthalmus pectinirostris, Periophthalmus magnuspinnatus, and Periophthalmus modestus) were clustered into a monophyletic clade. Within this clade, S. histophorus and B. pectinirostris were clustered into a sister clade, P. magnuspinnatus, and P. modestus clustered into a second sister clade with a 100% support rate for all nodes in these clades. This topology is also supported in other phylogenetic analyses (McCraney et al., 2020; Steppan et al., 2022). The divergence time analysis shows that Gobiiformes diverged from Kurtiformes approximately 96 Mya and S. histophorus diverged with B. pectinirostris approximately 30.9 Mya. Compared with the nearest ancestor, a total of 920 expanded and 1,594 contracted gene families were found in S. histophorus (Figure 2).
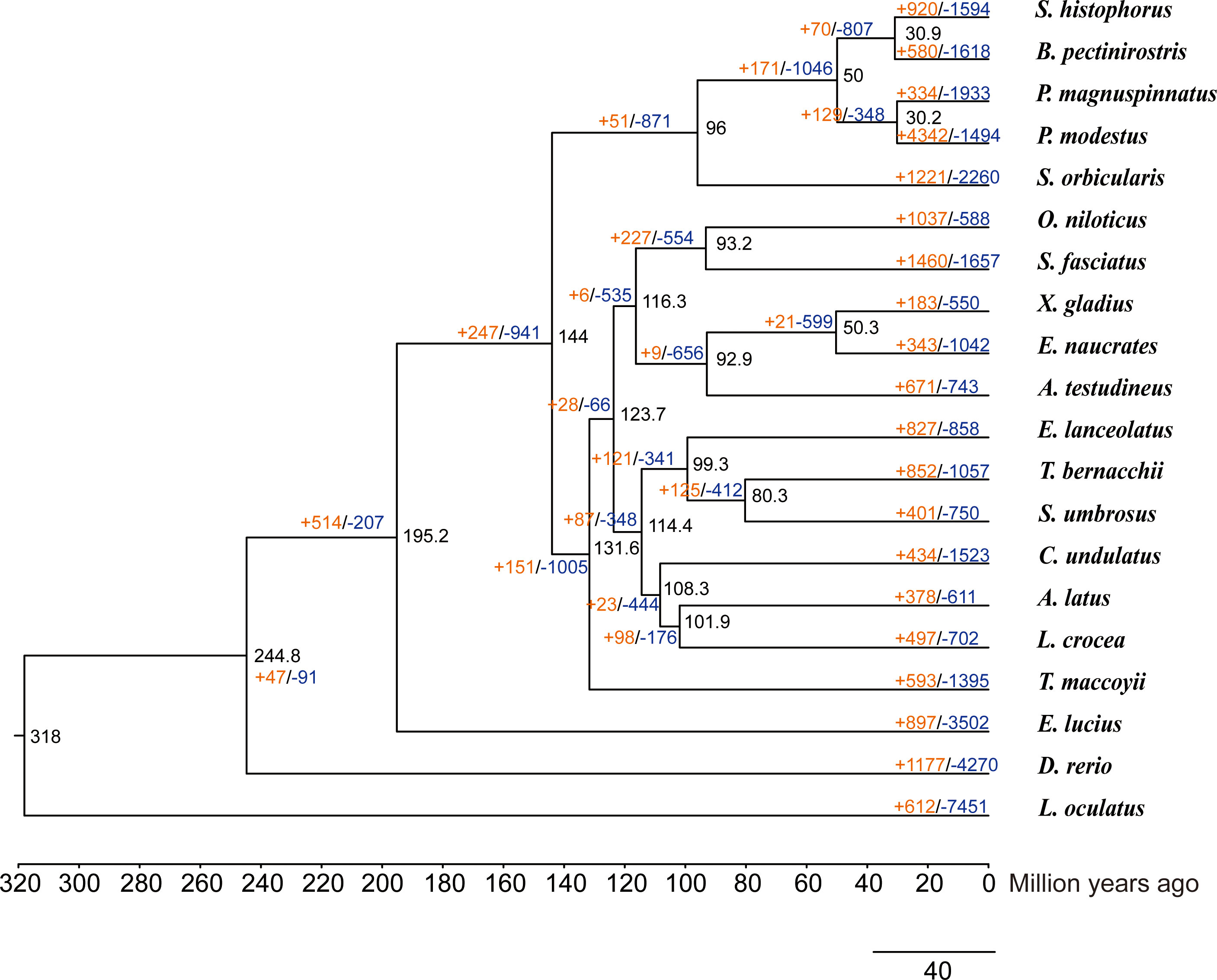
Figure 2 Phylogenetic tree of Scartelaos histophorus with 19 other species. Black numbers at nodes represent divergence time. The orange numbers and blue numbers represent the numbers of expanded and contracted gene families, respectively.
Materials and methods
Ethics approval statement
Sample collection was carried out in accordance with the approved guidelines of the Good Experimental Practices adopted by the Institute of Zoology, Chinese Academy of Sciences (CAS). All experimental procedures and sample collections were conducted under the supervision of the Committee for Animal Experiments of the Institute of Zoology, CAS.
Sample collection
We collected one male and one female Walking goby mudskipper from a beach in Xiapu County (26°52′N, 120°0′E), Fujian Province, China. The female muscle sample was used for genome sequencing, including Illumina, PacBio CCS, and Hi-C sequencing. To protect the integrity of the DNA, all samples were immediately frozen in liquid nitrogen for 20 min and then stored at -80°C. For RNA sequencing, we selected the brain, eye, gill filaments, heart, liver, pectoral fin, skin, and ovary of the female sample and testis of the male sample; all tissues were preserved in RNAlater solution and stored at -20°C. All samples were sent to Novogene (Tianjin, China) for sequencing.
For short-read sequencing, sequencing libraries were generated using the TruSeq Nano DNA HT Sample Preparation Kit (Illumina USA) following the manufacturer’s protocol. These libraries were sequenced by the Illumina NovaSeq 6000 platform, and 150-bp paired-end reads were generated with an insert size around 350 bp. For long-read sequencing, the sample DNA constructs circular consensus sequencing (CCS) libraries and sequences them using a PacBio Sequel platform. The Hi-C (high-throughput chromosome conformation capture) sequencing was performed as follows: muscle tissues were cross-linked by 4% formaldehyde solution to capture the interacting DNA segments, and chromatin was digested with the restriction enzyme MboI, sequenced on the Illumina HiSeq 2500 platform (PE 125 bp) (Lieberman-Aiden et al., 2009; Belton et al., 2012). For RNA sequencing, a paired-end RNA-sequencing library was constructed and 150-bp paired-end reads were produced on the Illumina NovaSeq platform.
Estimation of genome size
To estimate the genome size of S. histophorus, we used Illumina reads via kmerfreq (v. 4.0) (https://github.com/fanagislab/kmerfreq) to count k-mer frequency with a k-mer size of 17. Then, the genome size, repeat sequence percentage, and heterozygosity ratio were estimated by GCE (v. 1.0.2) (Liu et al., 2013).
Genome assembly and evaluation
The contig-level genome was assembled using PacBio HiFi reads by hifiasm (v. 0.16.1-r375) (Cheng et al., 2021) with default parameters. Next, to anchor the contigs into chromosomes, we aligned the Hi-C sequencing data into contigs using Juicer (v. 1.6) (Durand et al., 2016). The contigs were finally linked into 24 distinct chromosomes by 3D-DNA (v. 180922) (Dudchenko et al., 2017). A Hi-C contact matrix of the genome was visualized in Juicebox software and manually corrected in view of the chromosome interactions (Durand et al., 2016). BUSCO (v. 5.2.2) (Manni et al., 2021) was used with the library “vertebrata_odb10” and “actinopterygii_odb10” to evaluate the final genome completion. Illumina short reads were mapped onto the assembled genome used BWA (v. 0.7.17-r1188) (Li and Durbin, 2009) to evaluate completeness and accuracy of the genome.
Repeats and transposable element annotation
Repetitive sequences of the S. histophorus genome were annotated using a combination of ab initio and homology-based methods. First, LTR_FINDER (v. 1.07) (Xu and Wang, 2007) and RepeatModeler (v. 2.0.3) (http://www.repeatmasker.org/RepeatModeler.html) were used to construct an ab initio repeat library. The library and Repbase database (Bao et al., 2015) were then used to detect repetitive sequences by RepeatMasker (v. 4.1.2-p1) (http://www.repeatmasker.org/RepeatModeler.html).
Gene prediction and annotation
Protein-coding genes were annotated using three approaches, namely, homology-based, RNA-seq based, and ab initio prediction. For homology-based annotation, the protein-coding sequences of Larimichthys crocea, Lates calcarifer, Oreochromis niloticus, Anabas testudineus, Danio rerio, and Periophthalmus magnuspinnatus were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/assembly/), and the Periophthalmus modestus genome and annotation files were downloaded from http://gigadb.org/dataset/100957 (Yang et al., 2022). We retained only the longest transcript from each gene as a representative, and then BLAST was used to align the gene to the genome with an E-value cutoff of 1E-5. GeneWise (v. wise2-4-1) was used to predict gene structures based on the homology alignments (Birney et al., 2004). For RNA-seq-based annotation, the RNA-seq reads were assembled using Trinity (v. 2.13.2) (Grabherr et al., 2011), then the assembled transcripts and the genome with repeat sequences masked used PASApipeline (v. 2.5.2) (Haas et al., 2003) to generate the transcriptome evidence annotation. TransDecoder (v. 5.5.0) (https://github.com/TransDecoder/TransDecoder/wiki) was used to predict open reading frames. For de novo prediction, GeneMark (v. 4) (Lomsadze et al., 2005) and Augustus (v. 3.4.0) (Stanke et al., 2004) were used to generate ab initio predicted gene sets. In the end, EVidenceModeler (v. 1.1.1) (Haas et al., 2008) was used to integrate the results from the above three methods producing the final gene set. Circos software was used to visualize gene density, repeat-sequence density, GC content, and collinear gene blocks in the genome (Krzywinski et al., 2009). For gene functional annotation, InterProScan (v. 5.55-88.0) (Jones et al., 2014) was used to search in Pfam databases (Mistry et al., 2021) to annotate the functional domains of the gene set. In addition, each gene assigned by BLASTP against the Kyoto Encyclopedia of Genes and Genomes (KEGG), NR, and Swiss-Prot databases (Ogata et al., 1999) to get homology-based function assignments with an e-value cutoff of 1 × 10-5.
Comparative phylogenomics of S. histophorus
To clarify the evolutionary relationship between S. histophorus and other related species, we reconstructed a phylogeny with 19 species representing 16 orders. The calibration times were Lepisosteus oculatus–Danio rerio (295–334 Mya), Danio rerio–Esox Lucius (206–252 Mya), Thunnus maccoyii–Larimichthys crocea (106–144 Mya), Larimichthys crocea–Acanthopagrus latus (102–127 Mya), and Oreochromis niloticus–Salarias fasciatus (82–131 Mya), which query from TimeTree (http://www.timetree.org/). The detailed information about the species used for comparative phylogenomics is shown in Supplementary Table 10. A homologous gene set was clustered using OrthoFinder (v. 2.5.4) (Emms and Kelly, 2019) software with the msa method. Then, single-copy genes were identified and aligned with muscle. Merged alignment results were selected and used to identify the best-fit models of amino acid replacement using ProtTest (v. 3.4.2). The results show the best-fit models to be VT+F. The phylogenetic relationship among these species was estimated using RAxML (v. 8.2.12) (Stamatakis, 2014) with the VT+F models and 100 bootstrap. Divergence times were estimated by the MCMCtree within the PAML package (v. 4.9f) (Yang, 2007), and the reference divergence times were checked in the TimeTree database (Kumar et al., 2017). We measured the expansion and contraction of orthologous gene families based on the maximum likelihood tree using the software CAFÉ (v. 4.0.2) (De Bie et al., 2006). For a full list of programs and parameters used, see Supplementary Table 11.
Reuse potential
We present a chromosome-level high-quality genome assembly and annotation of S. histophorus using Illumina, PacBio HiFi, Hi-C, and RNA sequencing. The assembled genome is ~869.5 Mb, with contig N50 and scaffold N50 values of 9.02 and 35.28 Mb, respectively; based on multiple annotation strategies, a total of 24,223 protein-coding genes were annotated. Our phylogenomic analyses infer the evolutionary placement, divergence time, and gene family change of S. histophorus. This newly annotated genome can be a valuable resource for multiple avenues of the research on the adaptive evolution of transitions between aquatic and terrestrial environments. This is relevant not only within the mudskippers, with show multiple transition to a terrestrial lifestyle, but also in early tetrapods, across fishes (e.g., catfishes, killifishes, blennies), and other major clades (e.g., turtle/tortoise, Cetaceans) that exhibit this transition. Specifically, an annotated genome assembly allows for identification of candidate genes involved in these transitions.
Data availability statement
The raw reads and sequences of genome assembly and transcriptome sequence data generated in this study have been deposited in the National Genomics Data Center (NGDC) with accession number GWHBJEH00000000, and the genome assembly and annotation files are available at https://figshare.com/articles/dataset/A_Chromosome-level_genome_assembly_of_the_Walking_goby_Scartelaos_histophorus_/20174018.
Ethics statement
The animal study was reviewed and approved by Committee for Animal Experiments of the Institute of Zoology, CAS.
Author contributions
JZ supervised the study and managed the project. JZ, XL, and JQ collected the samples in-field. XL performed the genome sequencing, assembly, annotation, genetic data analyses, and writing of the draft manuscript. JQ, JBC, and JC contributed to the taxonomy and phylogeny issues. JZ, XL, and JBC finished the final manuscript with contributions from all other authors. All authors contributed to the data interpretation and manuscript revision and approved the submitted version.
Funding
The grants supporting this research were received from the State Oceanic Administration of China (JDXT 2018-03) and the Chinese Academy of Sciences, China (XDA19050202), both to JZ.
Acknowledgments
We express sincere thanks to Professors Hanlin Wu and Yong Ni for their long-term encouragement and support. We would like to especially thank Professor Ming Li for his technical assistance. The authors are grateful to Fude Chen, Huaikang Chen, Haoming Lu, and Minzheng Li for their kind help in the sample collections. Furthermore, special thanks are given to Yufan Mao (Jiangnan University, China), who provided a scientific illustration of the fish.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.966275/full#supplementary-material
References
Arai R. (2011). Fish karyotypes: A check list (Tokyo: Springer Tokyo Press). doi: 10.1007/978-4-431-53877-6
Bao W., Kojima K. K., Kohany O. (2015). Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6, 11. doi: 10.1186/s13100-015-0041-9
Belton J. M., McCord R. P., Gibcus J. H., Naumova N., Zhan Y., Dekker J. (2012). Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58 (3), 268–276. doi: 10.1016/j.ymeth.2012.05.001
Birney E., Clamp M., Durbin R. (2004). Genewise and genomewise. Genome Res. 14 (5), 988–995. doi: 10.1101/gr.1865504
Cheng H., Concepcion G. T., Feng X., Zhang H., Li H. (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18 (2), 170–175. doi: 10.1038/s41592-020-01056-5
Corush J. B., Pierson T. W., Shiao J.-C., Katayama Y., Zhang J., Fitzpatrick B. M. (2022). Amphibious mudskipper populations are genetically connected along coastlines, but differentiated across water. J. Biogeography 49(4), 767–779. doi: 10.1111/jbi.14345
De Bie T., Cristianini N., Demuth J. P., Hahn M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22 (10), 1269–1271. doi: 10.1093/bioinformatics/btl097
Dudchenko O., Batra S. S., Omer A. D., Nyquist S. K., Hoeger M., Durand N. C., et al. (2017). De novo assembly of the aedes aegypti genome using Hi-c yields chromosome-length scaffolds. Science 356 (6333), 92–95. doi: 10.1126/science.aal3327
Durand N. C., Robinson J. T., Shamim M. S., Machol I., Mesirov J. P., Lander E. S., et al. (2016). Juicebox provides a visualization system for hi-c contact maps with unlimited zoom. Cell Syst. 3 (1), 99–101. doi: 10.1016/j.cels.2015.07.012
Emms D. M., Kelly S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20 (1), 238. doi: 10.1186/s13059-019-1832-y
Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., et al. (2011). Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29 (7), 644–652. doi: 10.1038/nbt.1883
Haas B. J., Delcher A. L., Mount S. M., Wortman J. R., Smith R. K. Jr., Hannick L. I., et al. (2003). Improving the arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31 (19), 5654–5666. doi: 10.1093/nar/gkg770
Haas B. J., Salzberg S. L., Zhu W., Pertea M., Allen J. E., Orvis J., et al. (2008). Automated eukaryotic gene structure annotation using evidencemodeler and the program to assemble spliced alignments. Genome Biol. 9 (1), R7. doi: 10.1186/gb-2008-9-1-r7
Hu W., Mu Y., Lin F., Li X., Zhang J. (2022). New insight into visual adaptation in the mudskipper cornea: From morphology to the cornea-related COL8A2 Gene. Front. Ecol. Evol. 10, 871370. doi: 10.3389/fevo.2022.871370
Jones P., Binns D., Chang H. Y., Fraser M., Li W., McAnulla C., et al. (2014). InterProScan 5: Genome-scale protein function classification. Bioinformatics 30 (9), 1236–1240. doi: 10.1093/bioinformatics/btu031
Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., et al. (2009). Circos: An information aesthetic for comparative genomics. Genome Res. 19 (9), 1639–1645. doi: 10.1101/gr.092759.109
Kumar S., Stecher G., Suleski M., Hedges S. B. (2017). TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34 (7), 1812–1819. doi: 10.1093/molbev/msx116
Li H., Durbin R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25 (14), 1754–1760. doi: 10.1093/bioinformatics/btp324
Lieberman-Aiden E., van Berkum N. L., Williams L., Imakaev M., Ragoczy T., Telling A., et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326 (5950), 289–293. doi: 10.1126/science.1181369
Liu B., Shi Y., Yuan J., Hu X., Zhang H., Li N., et al. (2013). Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv preprint arXiv:1308.2012. doi: 10.48550/arXiv.1308.2012
Lomsadze A., Ter-Hovhannisyan V., Chernoff Y. O., Borodovsky M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33 (20), 6494–6506. doi: 10.1093/nar/gki937
Manni M., Berkeley M. R., Seppey M., Simão F. A., Zdobnov E. M. (2021). BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38 (10), 4647–4654. doi: 10.1093/molbev/msab199
McCraney W. T., Thacker C. E., Alfaro M. E. (2020). Supermatrix phylogeny resolves goby lineages and reveals unstable root of gobiaria. Mol. Phylogenet Evol. 151, 106862. doi: 10.1016/j.ympev.2020.106862
Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G. A., Sonnhammer E. L. L., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49 (D1), D412–d419. doi: 10.1093/nar/gkaa913
Nelson J. S., Grande T. C., Wilson M. V. (2016). Fishes of the world (Hoboken: John Wiley & Sons). doi: 10.1002/9781119174844
Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M. (1999). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27 (1), 29–34. doi: 10.1093/nar/27.1.29
Pace C. M., Gibb A. C. (2009). Mudskipper pectoral fin kinematics in aquatic and terrestrial environments. J. Exp. Biol. 212 (Pt 14), 2279–2286. doi: 10.1242/jeb.029041
Pan L., Zhang G., Wei K., Zhang J. (2010). A comparison of the gill morphometry of four mudskippers (gobiidae: oxudercinae) from southeast asia and oceania: With comments on ecological adaptation. Chin. J. Zoolog. 45 (04), 1–10. doi: 10.13859/j.cjz.2010.04.003
Patzner R. A., Van Tassell J. L., Kovacic M., Kapoor B. (2011). The biology of gobies (Boca Raton: CRC Press). doi: 10.1201/b11397
Sayer M. D. J. (2005). Adaptations of amphibious fish for surviving life out of water. Fish and Fisheries 6(3), 186–211. doi: 10.1111/j.1467-2979.2005.00193.x
Stamatakis A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 (9), 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke M., Steinkamp R., Waack S., Morgenstern B. (2004). AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 32 (Web Server issue), W309–W312. doi: 10.1093/nar/gkh379
Steppan S. J., Meyer A. A., Barrow L. N., Alhajeri B. H., Al-Zaidan A. S. Y., Gignac P. M., et al. (2022). Phylogenetics and the evolution of terrestriality in mudskippers (Gobiidae: Oxudercinae). Mol. Phylogenet. Evol. 169, 107416. doi: 10.1016/j.ympev.2022.107416
Wang L., Xu M., Liu B., Jiang T., Zhang S., Yang J. (2013). Experimental study on morphology and kinematics of mudskipper in amphibious environments in 2013 IEEE International Conference on Robotics and Biomimetics (ROBIO), 1095–1100. doi: 10.1109/ROBIO.2013.6739610
Xu Z., Wang H. (2007). LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Yang Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24 (8), 1586–1591. doi: 10.1093/molbev/msm088
Yang Y., Yoo J. Y., Baek S. H., Song H. Y., Jo S., Jung S. H., et al. (2022). Chromosome-level genome assembly of the shuttles hoppfish, periophthalmus modestus. Gigascience 11 (1), 1–8. doi: 10.1093/gigascience/giab089
You X., Bian C., Zan Q., Xu X., Liu X., Chen J., et al. (2014). Mudskipper genomes provide insights into the terrestrial adaptation of amphibious fishes. Nat. Commun. 5, 5594. doi: 10.1038/ncomms6594
You X., Sun M., Li J., Bian C., Chen J., Yi Y., et al. (2018). Mudskippers and their genetic adaptations to an amphibious lifestyle. Anim. (Basel) 8 (2), 24. doi: 10.3390/ani8020024
Zhang J. (2001). A comparative study on the morphological adaptations in oxudercine gobies to their terrestrial life (Nagasaki: Doctoral dissertation, Nagasaki University).
Zhang J., Taniguchi T., Takita T., Ali A. B. (2000). On the epidermal structure ofboleophthalmus andscartelaos mudskippers with reference to their adaptation to terrestrial life. Ichthyolog Res. 47 (3), 359–366. doi: 10.1007/BF02674263
Keywords: Scartelaos histophorus, Mudskipper, genome, water-to-land transition, amphibious lifestyle
Citation: Li X, Qi J, Corush JB, Chen J and Zhang J (2022) A chromosome-level genome assembly of the Walking goby (Scartelaos histophorus). Front. Mar. Sci. 9:966275. doi: 10.3389/fmars.2022.966275
Received: 10 June 2022; Accepted: 21 July 2022;
Published: 15 August 2022.
Edited by:
Jonathan Sandoval, Flinders University, AustraliaReviewed by:
Linlin Zhao, Ministry of Natural Resources, ChinaXiaowei Song, Xinyang Normal University, China
Copyright © 2022 Li, Qi, Corush, Chen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Zhang, zhangjie@ioz.ac.cn
†These authors have contributed equally to this work and share first authorship